講師コラム:長谷川 秀夫 先生
『 成分の定量分析法バリデーション
(Analytical Method Validation ) 』
測定方法や測定結果の「精確さ:accuracy」を確保するためには、測定値の信頼性を検証する「分析法バリデーション」、すなわち分析法に由来する誤差が原因で生じる試験結果について、その判定の誤りの確率が許容できる範囲であることを科学的に検証しなければならない。
化学分野で分析法バリデーションを実施する場合に要求される要素の指針として、次の項目が示されている。
1) 同定及び選択性・特異性の確認: Specificity
2) 感度・検出限界: Detection Limit
3) 定量限界 : Quantitative Limit
4) 検量線の実用範囲 : Range for calibration
5) 検量線の直線性: Linearity for calibration
6) 正確さ;真度/精度: Accuracy / Precision
7) 堅牢性(頑健性): Robustness
8) 回収率 : Recovery
講師コラムテーマ
第1回 バリデーションの要件1)同定及び選択性・特異性の確認: Specificity
伝統的技法に見られる特異的検出法;籾粒・玄米の分別;千石通し
第2回 バリデーションの要件2) 感度・検出限界: Detection Limit
電子天秤による計測では場所によって質量測定値は異なる。
第3回 バリデーションの要件3) 定量限界 : Quantitative Limit
単位;ダイヤモンドの大きさを示すカラットの起源
吸光度値、pHの値は対数表示:Logスケール
第4回 バリデーションの要件4) 検量線の実用範囲 : Range for calibration
MRI用造影剤の開発(時定数、Free induction decay)
第5回 バリデーションの要件5) 検量線の直線性: Linearity for calibration
低濃度溶液における溶媒の不純物
第6回 バリデーションの要件6) 正確さ;真度/精度: Accuracy / Precision
計測値の四捨五入処理は、合理的か?
(いろいろな選択股があることを知り柔軟に考える。;動物の習性事例)
第7回 バリデーションの要件7) 堅牢性(頑健性): Robustness
HPLCカラムは“生き物”;カラム内の環境は時間と伴に変化する。(再現性)
第8回 バリデーションの要件8) 回収率 : Recovery
食品標準成分表に見られるマグロの脂質含有量(規格及び試験法は一体である)
[1] [2] [3] [4] [5] [6] [7] [8]
第1回 [バリデーションの要件1)同定及び選択性・特異性の確認: Specificity
伝統的技法に見られる特異的検出法;籾粒・玄米の選別;千石通し](2009/8/18)
分析機器では、技術の開発・進展に伴い測定値がデジタル化され、さらに装置内部がブラックボックス化される中で高感度検出が可能となるにつれ、試料を入れれば何らかの測定値が得られるようになってきた。
その測定方法や測定結果の「精確さ:accuracy」を確保するためには、測定値の信頼性を検証する「分析法バリデーション」、すなわち分析法に由来する誤差が原因で生じる試験結果について、その判定の誤りの確率が許容できる範囲であることを科学的に検証しなければならない。
化学の分野では分析法バリデーションを実施する場合に要求される要素の一つとして、「選択性・特異性の確認: Specificity」が求められている。
元禄元年(1688年)ごろから使用されている「籾」と「玄米」、「玄米」と「玄米中のくず米」、「精米」と「精米中の砕け米」とをそれぞれ選別する農器具(図―1;「千石通し」、「万石通し」など)がある。
これらの伝統的な農器具では、篩の機能をさらに発展させ、網の穴サイズ、網の枚数や傾斜角度を変えることにより、「粒」の物性を巧みに生かして、「粒」を選択的・特異的に選別する技法が使用されている。

図―1;「籾」と「玄米」などを分別する農器具
米は収穫された「籾粒」から「玄米」を取り出し精米される。
したがって「玄米」は「籾粒」中に存在していたわけであるから、一般にはその両者を選別するには、その大小関係に注目し、篩を使用してサイズの差で選別する方法が考えられる。
しかし、図―2に見られるようにサイズの異なる「玄米」及び「籾粒」が存在する。

図―2;大きさの異なる「籾粒:paddy」と「玄米:brown rice」が存在する。
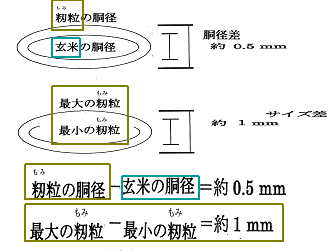
その「籾粒:paddy」と「玄米:brown rice」の平均的なサイズを調べてみる。
分別したい「籾粒」と「玄米」のそれぞれの胴径の平均的なサイズの差は、
籾粒の胴径―玄米の胴径=約0.5mmである。
一方、「籾粒」について見るとその大小のサイズの差は、
最大籾粒―最少籾粒=約1 mmもある。
このことは分別したい「籾粒」と「玄米」のサイズの差:約0.5mmに対して、「籾粒」自身の大小サイズの差が約1 mmもあるため、原理的に「籾粒」と「玄米」をサイズの差では分別できないことを意味する。
図―3;分別したい「籾粒」と「玄米」の平均的なサイズの差
そこで「籾粒」と「玄米」とを選別する農器具(「千石通し」、「万石通し」など)では、「籾粒」と「玄米」の比重と摩擦係数の差に注目して、両者の選択性を向上させ実用化させている。

図―4;デジタルカメラ;RICOH GX200による直接撮影
著者の感想;
この写真は、デジタルカメラで直接撮影したものである。
驚いたことに籾粒の“ひげ“まで鮮明に撮影されていることである。
デジカメは焦点深度が深いのでこの様な撮影が可能となった。
このことは顕微鏡による撮影に頼ることなく研究開発の記録にデジカメが活用できるのではないでしょうか。
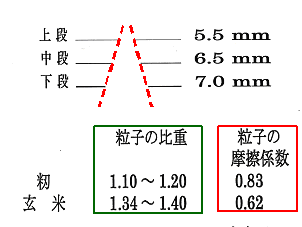
図―5; 分別したい「籾粒」と「玄米」の物性;粒子の比重・摩擦係数
図―4の写真に見られるように「籾粒」の表面には、“とげ”があるために「籾粒」の摩擦係数0.83は「玄米」0.62に比べて大きく、また、「籾粒:1.10~1.20」は「玄米;1.34~1.40」と比べて比重が小さい(軽い)。
これら物理的特性を生かして、これらの農器具では両者の選別を粒子の比重及び摩擦係数の差により選別されている。
ここで篩の役割は、比重が小さく(軽い)、摩擦係数の大きい「籾粒」の篩上の滞留時間を長くする役目を果たしている。したがって複数使用されている篩の網の穴のサイズは上段ほど小さくなっている。サイズの大小で選別するならば上段ほど大きな穴の網が使用されるはずである。
近年、分析機器の検出器には、多くの種類がある。
HPLCの検出器と例としては、次の様なものがある。
・吸光光度検出器( Ultraviolet-Visible detector:UV/VIS )
・蛍光光度検出器( Fluorescence detector :LD )
・示差屈折率検出器( Refractive Index detector:RI )
・電気化学検出器 ( Electrochemical Detector:ECD )
・電気伝導度検出器( Electric conductance detector:CD )
・質量分析器( Mass Spectrograph:MS )
・赤外分光検出器( Infrared detector:IR )
・気化光散乱検出器( Evaporative Light Scattering Detector:ELSD )
・荷電化粒子検出器(Charged Aerosol Detector:CAD )
これらの検出器を選択する際には、目的とする成分をより選択的・特異的に、しかもより高い検出感度で検出できるものが選ばれる。
定量分析で最も重要な要件は、まず目的とする成分に対する選択性・特異性が確保されているか否かを確認しなければならない。
一般に製剤の分析では、「有効成分を含有していないブランクの疑似試料」が調製できるので「同定及び選択性・特異性の確認: Specificity」は容易である。
しかし、生体成分、食品などの天然物では、目的とする成分が存在しない「ブランクの疑似試料」が調製できない。
そこで牛乳及び乳製品中のビタミンK定量法として、ビタミンK検出に対する選択性・特異性を確認する分析方法を紹介しよう。
事例; 検出器の選択・組み合わせ及び 測定条件の設定方法の事例
選択性・特異性の向上;食品中のビタミンKの分別定量と定量値の評価
長谷川秀夫,ビタミン,71,(8),365 (1997)
検出器 ;蛍光検出器と電気化学的検出器
測定方法 ;HPLCカラムで分離・精製した後,蛍光光度法及び電気化学的検出(ECD:Electrical Chemical Detection)法で測定する。
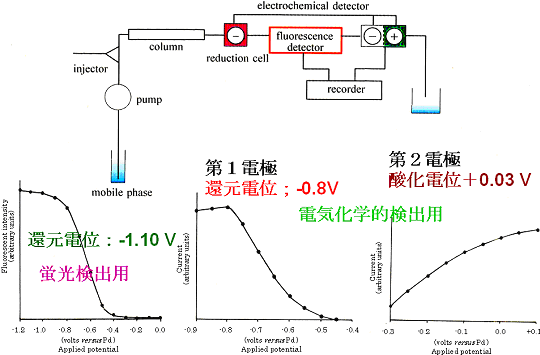
図―6 分析用HPLCシステムのダイヤグラム(各ユニットの配置図)
及び測定電位設定のための還元電位
―蛍光強度,酸化―還元電位,還元-酸化電位曲線,
ビタミンK1・ビタミンK2 混合標準溶液及び生乳試料溶液について,蛍光及びECD両検出器から得られた代表的なクロマトグラムを図―7に示した。
各検量線用の標準溶液及び生乳試料についてビタミンK1及びビタミンK2各測定値並びに両測定値の比をまとめて表1-1(a)及び表1-1(b)にそれぞれ示した。
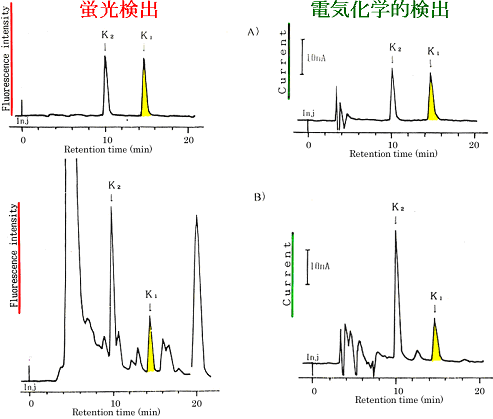
図7 ビタミンK1・ビタミンK2 混合標準溶液及び生乳試料溶液を蛍光検出器
及びECD検出器から同時に得られた代表的なクロマトグラム
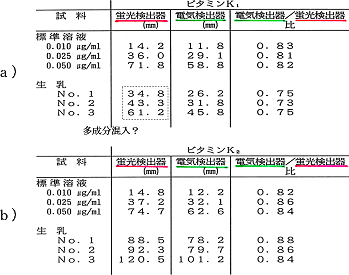
表ー1 検量線用の標準溶液及び生乳試料について蛍光検出器及びECD検出器から同時に
得られたビタミンK1及びビタミンK2各測定値並びに両測定値の比
ビタミンK1の測定結果(表-1(a))より,各検量線用の標準溶液及び生乳試料について,蛍光出及びECDにより得られたそれぞれの測定値の比を比較する。標準溶液の平均値:0.83に対して試料溶液は,0.74であり明らかな差が認められる。したがって,蛍光検出器から得られたビタミンK1のクロマトグラムのピークから,生乳試料には,他の成分が混入していると推定することができる。
一方,ビタミンK2に対する測定結果(表-1(b))では,標準溶液の平均値:0.84に対して試料溶液の場合は,0.86である。両測定値とも計測誤差内で一致しているため,生乳試料の場合には,この両測定値の比率より設定した測定条件は妥当なものと推定される。また,両測定値とも信頼性ある定量値として評価することができる。
定量分析の重要な要件である目的物質に対する選択的・特異的な検出法を設定する際には、まずは各成分の選択性の向上にあるわけですから検体により条件は異なるため、既存の方法に捉われることなく分析の目的を明確にすることにより、効果的な定量方法を選択したいものです。
[topへ]
第2回 バリデーションの要件2) 感度・検出限界: Detection Limit
(2009/9/1)
質量(mass)は物体固有の物理量で同じ物体である限り不変であるが、一般に使用されている電子天秤によって測定される重さ(重力質量:gravitation mass)の値は場所によって異なる。
電子天秤は重力質量を測定しているために、その測定値は場所(重力加速度の違い)により、あるいは高度差(浮力)により異なる。しかし古くから使用されていた天秤(バランス)による方法での計測では、天秤方式による比較測定であるからこの様なことを考慮する必要はなかった。
このように計測・分析装置を使用する場合には、分析法バリデーションが必要となるのである。
測定と計測の違い (矢野 宏;誤差を科学する Blue Backs 1994より引用)
計測とは、測定の結果を有効に活用するということを含む仕事
{測定}が観察データであるとすれば、{計測}はもの作りのように、目的を持ったデータをとらなければならない。
分析によって得られた計測値は、目的を持ったデータであるから商品設計、研究・開発、品質管理などに使用され、ある時は法律・規制の対象にもなり、また国際問題にもなりかねない。さらに計測された値は、数値としてデジタル化され歩きだし、そして走り出す。
測定方法や測定結果の「精確さ:accuracy」を確保するためには、測定値の信頼性を検証する「分析法バリデーション」、すなわち分析法に由来する誤差が原因で生じる試験結果について、その判定の誤りの確率が許容できる範囲であることを科学的に検証しなければならない。
バリデーション:Validationとは;科学的根拠、妥当性の確認の意
期待されている結果を検査する作業
分析法バリデーションの目的:Analysis validation
試験に用いる分析法が使用される意図にふさわしいことを立証することにある。
したがって一律に適用するものではなく、「Case by case の許容範囲」で適用される。
「Case by case の許容範囲」と言ってもやはり目安となる数値があると便利である。
指針として示されている幾つかの事例を下記に示すがあくまでも目安であり、試験に用いる分析法及び測定対象物質により異なることは言うまでもない。
感度・検出限界:Detection limit
試料中に存在する分析対象物の最低検出可能な量。
< 感度・検出限界評価方法の目安 >
測定値が正規分布し連続な場合には、測定対象物質を用いて検出限界付近の濃度領域で検量線を作成し、測定値の標準偏差:σ(シグマ)と検出限界付近のブランク試験(濃度ゼロ)における検量線の傾き:a より求める。
検出限界=3.3σ/a
< 検出限界設定の目安 >
一般的には、信号の高さ/雑音の高さの比が2~3を目安として検出限界設定されている。
設定した分析法の検出限界値が設定された規格値以下であることを確認する。
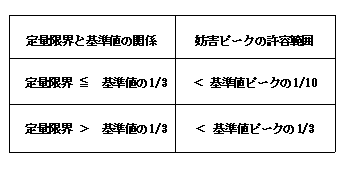
< 定量限界及び基準値の比と妨害ピークの許容範囲の目安 >
[topへ]
第3回 バリデーションの要件3) 定量限界 : Quantitative Limit
単位;ダイヤモンドの大きさを示すカラットの起源
吸光度値、pHの値は対数表示:Logスケール(2009/9/15)
1.最少単位
ダイヤモンドは、古き時代より金よりも貴重な物質として扱われてきたが、金と異なり溶融してサイズを変形することが出来ない。
そこで宝石の大きさ;重さを表す最少単位としてイナゴマメ(図-1)の実(図-2)が使用された。
イナゴマメの実は、いずれもほぼ同一の重さ(約0.2 g)で堅く変形しにくい。その特性を生かしてイナゴマメの実が定量限界の最少単位としてダイヤモンドの重さの計量に使用されたのではないだろうか。
現在の宝石の質量単位:カラット(carat)の語源は、このイナゴマメ(Kerationギリシヤ語)とされている。 1907年のメートル条約会議で1カラットは200mgと定められた。

図-1;イナゴマメの木
(シチリア島;コンコルディア神殿付近で撮影)

図-2;イナゴマメ(Kerationギリシヤ語)の実

図-3;宝石の質量単位(カラット)の語源となったイナゴマメ(Kerationギリシヤ語)
の実と1円硬貨との比較(重さ・サイズ)

図-4;1円硬貨と米国1セント硬貨のサイズ及び重さ
各国で使用されている硬貨のサイズ及び重さは、それぞれの国で使用されている単位スケールのサイズ及び重さが採用されている。
例えば、
日本はメートル法であるから
1円硬貨の重さは1 g、
サイズは20 mm
米国では重さの単位としてポンド(pound)-オンス(ounce)、
長さの単位としてマイル(mile) - フイート(feet) - インチ(inch)が使用されているので
1セント( cent)の重さは、金衡1 pound x 1/12 = 1 ounce = 31.1 gの1/10 = 3.1 g、
サイズは、 1 inch = 25.4 mm x 3/4 = 19.0 mm
硬貨は正確に製造されているので家庭内で重さや長さを計測する場合の計量標準として使用できる。
2. 理化学領域で使用される計測スケールの対数:Log
1/100 、1/10、 10倍、100倍、 1,000倍単位のスケールを表現する場合、対数:Log 表示は大変便利である。
対数:Log表示値は十進法表示とは意味が異なることを理解しなければならない。例えばpHの1の差は対数:Logスケールであるから水素イオン濃度で10倍又は1/10の濃度差に相当することを意味する。
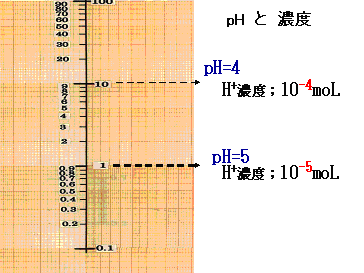
図-5;pH値と濃度のスケール
水素イオン濃度のpHスケールと同様に光吸収測定に使用される吸光度値も 対数:Log スケールである。
特に注意しなければならないのは、
・吸光度値:0.2以下の場合
試料セルの表面反射光、試料溶液内の散乱光等の影響が無視できなくなる。
・吸光度値:0.7以上の場合
吸光度値は対数表示値ゆえに吸光度差:1は、濃度換算で10倍に相当するため
吸光値が高い水準ではわずかな濃度変化が計測できない。
したがって吸光度値による定量する場合には、吸光度値0.2~ 0.7の濃度範囲での計測が推奨される。
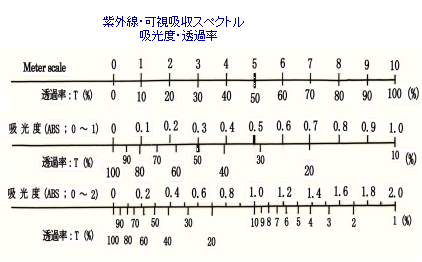
図-6;光吸収スペクトルにおける吸光度、透過率及びメータのスケール
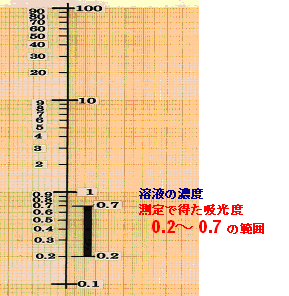
図-7;吸光度と濃度のスケール
液体クロマトグラムは吸光度の経時変化を描いている。
すなわちクロマトグラムは、設定された測定波長における溶出成分に基づく吸光度の変化量を測定している。したって吸光度定量法で推奨される吸光度値0.2~ 0.7の濃度範囲にこだわることはない。
しかし、HPLC の移動相の吸光度が著しく高い場合には、HPLC のベースラインが高いレベルで測定しているわけであるから、吸光度値の変化量の直線性が得られない(飽和現象)場合がある。HPLC は設定された波長における吸光度値の変化を測定するわけであるからベースラインの吸光度値が低いほど良好な測定条件となる。
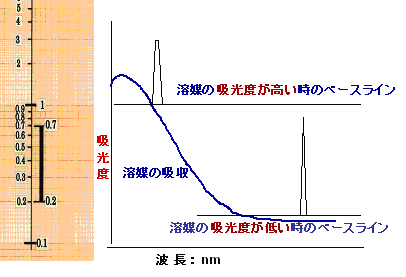
図-8;HPLC 測定における溶媒の吸光度と吸光度測定
3. バリデーションの要件;定量限界:Quantitative limit
試料中の分析対象物を適切な精度と真度で定量できる最低の量を定める。
<定量限界設定方法の事例>
測定値が正規分布し連続な場合
測定対象物質を用いて,定量限界付近の濃度領域で検量線を作成する。その回帰直線の残差(回帰方程式から得られた予測値と測定値の差)の標準偏差または回帰直線から推定したブランク試験(濃度ゼロ)における測定値の標準偏差をσ(シグマ)として,定量限界付近の検量線の傾き:aより定量限界を求める。
定量限界=10σ/a
<定量限界設定の目安>
一般的には,信号の高さ/雑音の高さ比=10を目安として定量限界が設定されている。また測定値の精度は,相対標準偏差で10%付近が許容される範囲である。
[topへ]
第4回 バリデーションの要件4) 検量線の実用範囲 : Range for calibration
MRI用造影剤の開発(時定数、Free induction decay)(2009/9/29)
1.絶対検量線法(Absolute Calibration Method)
定量分析の際に最も一般的に採用されている比較定量法で、純度既知の標準物質によって調整された濃度の異なる標準溶液を複数準備し、標準物質の質量と信号強度との関係(検量線 図-1)を作成する。
これを使って、標準溶液と試料溶液から得られた信号強度(ピークの高さ又は面積)を比較する方法で定量する。
検量線の実用範囲:Rangeとは、
適切な精度,真度及び直線性が得られる分析対象物の濃度範囲(上限及び下限の濃度:量)。
<実用範囲設定の目安>
実用的範囲;設定された規格値の±20%程度
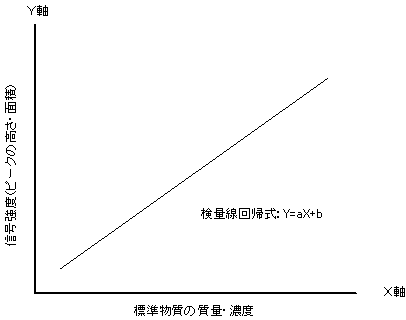
図-1 絶対検量線法
2.計測器で得られる信号の特性; Free induction decay・時定数
分析機器は、技術の開発・進展に伴い測定値がデジタル化され、さらに装置内部がブラックボックス化されてきた。そして高感度検出が可能となり、試料を入れれば何らかの測定値が得られるようになってきた。
そこで定量分析にされる測定値、すなわち分析機器で測定され表示される数値が得られる前段階の信号強度について振り返って見よう。
計測器で得られる信号は電気回路の特性から図-2のような曲線を描いて増大し、図-3のような曲線を描いて減衰する。したがって横軸を時間とすると厳密には無限大の時間後に前者の場合の信号は100 %強度になり、また後者の場合は無限大の時間経過後に信号は0 % 強度になる。
そこで信号は、システムの変化がそれぞれ1-1/e≒0.63 (63%)及び 1/e≒0.37(37%)に達した時間を時定数:Time constantとして処理される。
(立ち上がり(0%)点の傾斜のまま最終点(100%)まで到達したと仮定した時間)
一般に計測装置では測定条件として時定数を設定する。
時定数の設定は、信号波形のシャープ性、計測の横軸に相当する走引時間と関係し、信号のS(信号)/N(雑音)比を決定する因子ともなる。
一般の分析装置においては、時定数設定の表示として相対的な表現の「fast」、「Mid」、「slow」などの記号で示されているものもある。
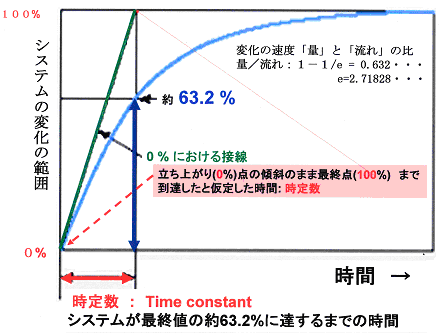
図-2 信号の変化が1-1/e≒0.63 (63%)に増大した時点(時定数)
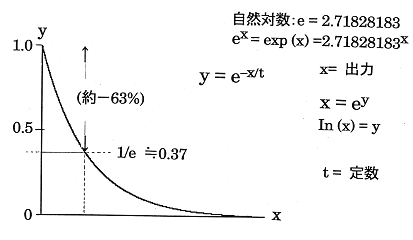
図-3 システムの変化が1/e≒0.37(37%)の水準に減衰した時点(時定数)
3.実用的な濃度範囲
医療用画像診断法として超音波、X-CT、MRI(Magnetic Resonance Imaging)法がある。
MRI法は、測定原理としてNMR(Nuclear Magnetic Resonance:核磁気共鳴)法が採用されていることから形態観察するだけでなく生化学的な情報が得られる特長がある。
その医療診断では、通常T1強調画像及びT2強調画像の2種類で診断が行われている。MRI信号強度(信号強度が強い;より白く画像化される場合)は、表-1に示す式のように生体を構成する水(水素核)のT1緩和時間及びT2緩和時間と関係がある。
両緩和時間は独立した現象の時間ではないために診断の目的に応じてT1を強調する測定条件又はT2を強調する測定条件で測定することにより、それぞれT1強調画像及びT2強調画像を得る。
表-1

T1強調画像及びT2強調画像におけるMRI強度と造影剤成分の濃度とは図-4に示す関係がある。したがってMRI用の造影剤では、定量分析に使用される検量線の実用範囲と異なり、実用的な機能を示す濃度領域がある(Pat.2911674)。
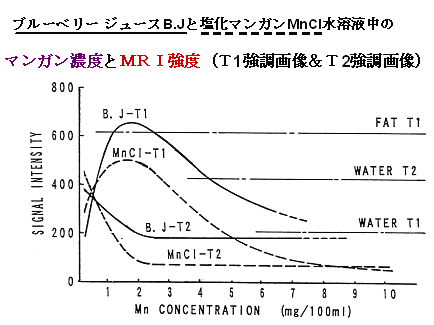
図-4 造影剤成分のマンガン:Mn+2濃度に対するMRI信号強度の関係
Jpn.J.Radiol.Technol.,12(2),302-312,1994
[topへ]
第5回 バリデーションの要件5) 検量線の直線性: Linearity for calibration
低濃度溶液における溶媒のJIS規格内の不純物(2009/10/13)
前第4回『バリデーションの要件4)』の検量線の実用範囲内で、標準溶液中の分析対象物の濃度(量)と測定値との関係の直線性を確認する。
< 事例;直線性評価方法 >
少なくとも濃度の異なる5 水準の標準溶液を用いて最小二乗法などの統計的手法を用いて,
その回帰式及び相関係数から直線性を評価する。
“ 検量線はほぼ直線性を示すけれども・・・・ 標準溶液調整用の希釈溶媒が不適当であったために・・・・・・・。”
事例; α-トコフェロール(α-tocopherol)の定量の際,標準溶液の検量線はほぼ直線となったが,
原点を通らず,回帰式グラフの切片が図-1のように,大きくマイナス(-)となった。
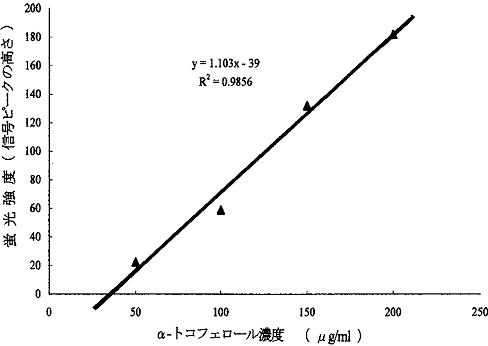
図-1 回帰式グラフの切片がマイナス(-)値を示した検量線
この事例における検量線用標準溶液の調整処方を表-1に示す。
表-1 検量線用の一連の標準溶液調製事例(調製された標準液の濃度)

ここでは標準品であるα-トコフェロールをn-ヘキサンで希釈し,検量線用の濃度の異なるそれぞれの溶液を調整している。ここで標準溶液調整時の標準原液量と溶媒(ヘキサン)量に着目してほしい。
α-トコフェロールを成分とする原液0.5 ml,1.0 ml,1.5 ml,2.0 mlに対し,n-ヘキサンを希釈溶媒として,それぞれ9.5 ml,9.0 ml,8.5 ml,8.0 mlを加える方法でいずれの標準溶液も10 mlになるように調整している。
この時,標準原液量と希釈溶媒量は,量的に逆比例の関係があるので標準原液の量が少ないほど加えられる希釈溶媒量の割合が多くなる(図-2)。すなわちn-ヘキサンに不純物成分として混在していた酸化剤成分によって消費されるα-トコフェロールの量は,一定の割合となるため,検量線の直線性は維持されることになる(図-1)。
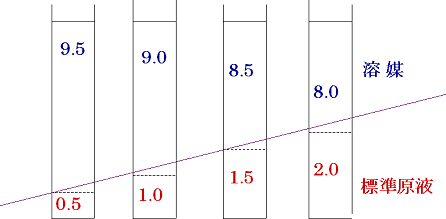
図-2 検量線用の一連の標準溶液調製事例
回帰式グラフのY軸切片がマイナスになったこの事例は,標準原液を希釈する溶媒:n-ヘキサン(n-hexane)に混在していた不純物が原因であった。
このα-トコフェロールはビタミンEの一種で抗酸化成分として機能し,酸化されやすい。
この事例が示すように,検量線用標準溶液を調整する場合,その濃度が極めて低い水準にあるために成分に対する保存条件及び溶媒に混在する不純物についても細心の注意が必要である。この様な事例からも少なくとも濃度の異なる5 水準の標準溶液を用いた検量線の必要性が提唱される。
一般的な注意事項
(1) 試薬の規格と不純物
「試薬の不純物は製造ロットにより異なる」ことを理解して使用する!
JIS規格特級,1級試薬などの不純物は,それぞれの規格内の不純物については保証されているものの,規格以外の成分については保証されていない。したがって妨害不純物の成分及び量が試薬の製造ロットにより異なるために分析の目的によっては、製造ロットごとに試薬による影響は異なる。
表-2 JIS規格 特級 ヘキサンの表示事例

(2) 定量分析の基本となる条件「標準溶液と試料溶液のマトリックスを酷似させる」を厳守する。
(3) 標準溶液調整の際,操作中の酸化による影響を考慮しなければならない場合、あらかじめ溶媒に抗酸化剤を添加しておく。
注)不純物としての酸化剤成分が混在した溶媒を使用した場合,酸化剤成分によって標準品の成分が
酸化されるため,調整した標準原液の濃度の力価は当然低下する。しかし,一般に標準原液は
成分が高濃度に調整されており,それに比べて溶媒に存在する酸化剤の量は極めて少ない。
したがって不純物由来の酸化成分による高濃度の標準原液に対する濃度への影響は少ない。
(4) 抗酸化成分を定量する際には,あらかじめ試料溶液及び標準原液にも強い抗酸化剤を添加し,酸化防止対策を施した環境下で試料を調製する。
この様な方法で溶媒中に存在する酸化剤成分や調製操作中の酸化による影響は無視できる場合もある。
ただし常に「標準溶液と試料溶液のマトリックスを酷似させる基本条件」の原則を考慮する。
< 提 案 ; 所要時間の短縮と同時的バリデーション >
濃度の異なる5 水準の標準溶液を用いた検量線により定量する場合、試験・検査時の所要時間が長くなる。しかし下記の「同時的バリデーションの実務的事例」のような方法を採用すれば測定所要時間も短縮され合理的な同時的バリデーションともなるのではないでしょうか。
検量線(事例;1ng,2ng,3ng,4ng,5ng/mL)の作成の後、安定性試験用の標準溶液(3ng/mL)の測定を試験・検査溶液(事例;A,B,C,D.E,F 試料溶液)の測定の途中に測定する。
この測定手順をとれば、検量線の評価(回帰式&相関係数)により安定性も確認され、さらに標準溶液(3ng/mL)を試料溶液の測定の途中に測定する方法で、測定の安定性及び測定条件の異常が確認できる。
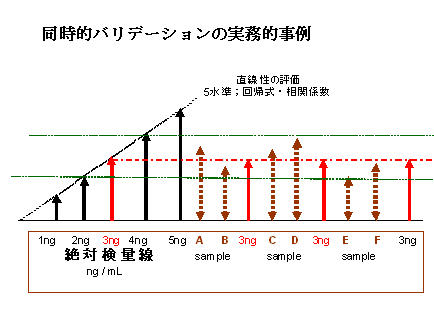
図-3 所要時間の短縮と同時的バリデーションの実施事例
[topへ]
第6回 バリデーションの要件6) 正確さ;真度/精度: Accuracy / Precision
計測値の四捨五入処理は、合理的か?
(いろいろな選択股があることを知り柔軟に考える。;動物の習性事例)(2009/10/27)
測定値の信頼性の確保
1. 「精確さ:accuracy」の評価
測定値の信頼性に相当する「精確さ:accuracy」は、個々の測定結果と採択された参照値(標準品としての値)との一致の程度で表される。それは、かたよりの成分(各測定結果に共通する系統誤差)と「ばらつきの成分」(不確定誤差)の両成分で構成される。換言すれば、[ 図-1 ]に示す「真度」と「精度」を総合的に表したものである。
図-1「精確さ:accuracy」評価の概念図
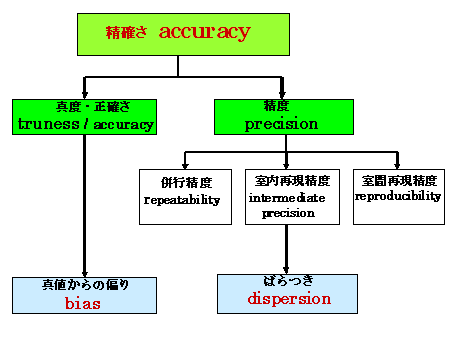
近年「精確さ」を評価する指標として、真の値が存在するはずである測定値の周りの範囲を意味する「不確かさ:uncertainty」という概念が用いられるようになってきた。
1.1. 真度(正確さ)(Trueness(Accuracy))
一連の多数の測定結果から得られる平均値と、採択された参照値(真の値)との間の一致の程度を示す。例えば、真の値を厳密には知ることできなくても、適切な標準物質あるいは既知試料の特定の参照値と測定値とを比較する。一般には、真の値からの「かたより:bias」の小さい程度として表現される。なお、本概念を正確さ(accuracy)と呼ぶ場合もあるが、最近では真度(trueness)が用いられる。
分析法の真度の推定値は,下記の2) 室内再現性または3) 室間再現性を求める時に得られる測定値の総平均と真の値との差として表される。
<評価の目安>
得られた真度の推定値と室内再現性または室間再現性の精度から計算される標準誤差の値から,
真度の95%信頼区間を計算する。この区間が「0」を含んでいることを確認するか,または同区間の
上限値及び下限値が分析法に要求される真度が基準の値の範囲内であることを確認する。
1) 併行精度(Repeatability)(Intra-assay)
試験室、試験者、装置、器具、及び試薬の製造ロットなどを変えることなく同一条件下で、
均一検体から採取した複数の試料を短時間内に繰り返し測定する場合の精度。
<液クロの場合の評価の目安>
液クロの保持時間に関する繰り返し測定の“ばらつき”の許容範囲として「3%以下」の数値が
提案されている。
2) 室内再現精度(Intermediate Precision)
同一試験室内で、試験日時、試験実施者、器具、機器、試薬製造ロット等の一部又はこれら全項目の
測定条件を変えて、均一でしかも安定性のある検体から採取した複数の試料を繰り返し測定した場合の
精度。
3) 室間再現精度(Reproducibility)
異なった施設間で測定する場合の精度。
1.2. 精度(Precision)
同一試料に対して定められた条件下で繰り返し測定された独立した測定値間の一致の「バラツキ」の程度を示す。一般には、測定値の分散、標準偏差又は相対標準偏差で表される。
2. 測定された測定値の単位から精確さ:accuracyの程度を把握・実感しておこう。
東京日本橋には国道の起点となる「道路元票」がある(図-2)。
そこから国道経由で下関市までは1,076 kmと表示されている。
他方、JRによる鉄道によると営業距離で約1,000 kmは、東京駅より山口県・防府駅付近に相当する。

ここで相対的数値として表わされるppmあるいはppbの単位について、長さ:距離の単位で示してみよう。
国道経由で東京から下関までの約1,000kmを測定した場合を考えてみる。
・1,000 kmに対してその 1 ppm は、1 m に相当する。
・1,000 kmに対してその 1 ppb は、1 mm に相当する。
このような単位で表現することによって、特に低濃度の成分の分析精度については、測定誤差を含む有効数字を考慮して表示しなければならないことが実感できる。
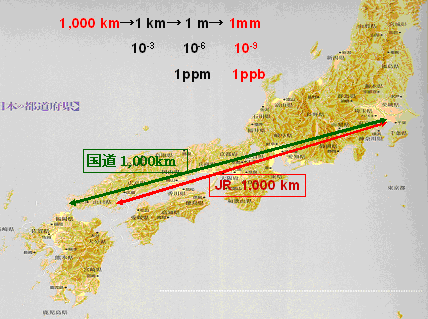
図-4 東京から1,000 kmの距離
3. 求められる計測精度
水泳競技で使用されるプールの国際公認コースの精度 は、どの程度なのであろうかと調べてみた。

国際公認コースの精度: 50 mの距離に対し、誤差 -0 mm、+10 mm以内
中心点を仮定するとその相対誤差は、約10/2 mm / 50,000mm = 100 ppm の精度が要求されていることになる。
4. 教科書的知識に対する先入観の再考
4以下を切り捨て、5以上を切り上げるいわゆる「四捨五入」の処理を行うとその確率は大きめに片寄り、確率的に1/2の割合で選択したことにならない。
例えば3.0, 3.1, 3.2, 3.3, 3.4, 3.5, 3.6, 3.7, 3.8, 3.9の10個の測定値について、小数点第1位を「四捨五入」する場合は、3.0は「四捨五入」の対象にならないので3.1~3.9の9個の測定値が対象となる。この時の「四捨五入」の処理では3.1~3.4の4点の測定値が切り捨てられ、3.5~3.9の5点の測定値が切り上げられることになる。

測定値の計算処理においてはこの様な問題点もあるため、日本工業規格(JIS)では十進法の数値の丸め方について「数値の丸め方 : Guide to the rounding of numbers : Z 8401」の規定がある。したがって測定された測定値は、有効数字を含め、どの様な計算方法で処理されたか明記する必要がある。特にコンピュターのソフトを利用して計算した場合には計算処理方法の記載が必須となる。
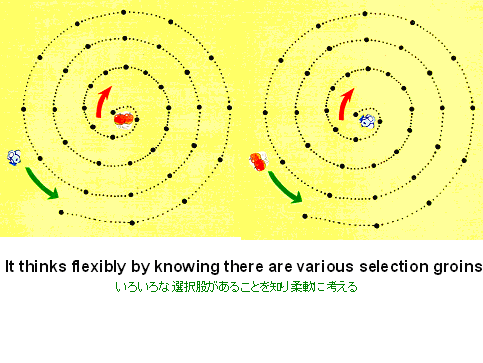
5. 取得した知識は動物的本能の中で構成されている
下記の左図のように金網で見通しの効く渦巻き状の通路を設け、その輪外にネズミを置き、その中心部に餌(赤色リンゴ)を置くと、そのネズミは中央の餌にありつく。
しかし逆に餌を外に置き、ネズミを中心に入れると餌にありつくことが出来ない。
何故か?
それは、前者の場合は、ネズミはネットを通して常に中心の餌を直視しながら接近できるので餌にありつける。
後者の場合、中央のネズミが外部の餌を目指すには、学習能力を取得して、一度、後ろ向きとなって餌から目を離なすために学習能力を必要とする。
したがって餌を外側に置いた場合には、中心に入れられたネズミを学習させなければ餌にありつけないのである。
この行為の本質は、1つのことを進める時にはそれに集中し、周りが見えなくなることが動物的な本能としてあることを物語っています。
試験・分析の方法の選択、管理・教育システムの構築方法などの場合においても、既存の方法に捕らわれることなく、情報交換等によりいろいろな選択股があることを知り、幅広く柔軟な考え方で効率的に分析精度の向上を目指したいものです。
[topへ]
第7回 バリデーションの要件7) 堅牢性(頑健性): Robustness
(2009/11/10)
1. 堅牢性〈頑健性〉:Robustness
分析法の信頼性を示す条件の1つ。
分析方法の条件について、想定される範囲内で測定の条件を故意に変動させたときに測定値が影響を受けない条件の範囲。
<堅牢性確認項目の事例>
分析法に共通する変動因子
ⅰ) 試験溶液の安定性(保存期間)
ⅱ) 試験溶液調整時の抽出時間
HPLCにおける代表的な変動因子
ⅰ) 移動相のpHによる変動の影響
ⅱ) 移動相の組成(試薬の製造ロット差など含む)の変動の影響
ⅲ) HPLCカラムの製造ロット差、メーカー差など
ⅳ) 測定温度
ⅴ) 移動相の流速
2. 液クロカラムは、“生き物”に例えられる中で信頼性ある定量値を如何にして得るか?
高速液体クロマトグラフィ(High Performance Liquid Chromatography;HPLC)、通称液クロは、溶液(移動相)を固定相の間に通すことにより、移動相と固定相間での成分の分配、吸着・脱離(吸脱着)現象によって、溶液中の各成分を分離・精製する技法である。
検体に対する前処理の簡便さと、注入から定量までが装置の組み合わせによって、自動的に行うことができるため、分析化学を始め多くの分野で使われている。
ところが、液クロは基本的な問題を抱えている。それは、移動相と固定相間での成分の分配、吸着・脱離(吸脱着)現象が環境の影響を受けやすく、測定ごと、さらに時間の経過とともに、カラム内での状態が刻々と変化するからである。この変化そのものが、液クロの特徴であり また条件を変化させることにより高い分離度で成分を分離・精製できることに繋がる。ゆえに“生き物”に例えられる。この本質の一つは、分離カラムのベースとなっているシリカゲル自身の構造特性に由来するとも考えられる。検体溶液中には、通常多くの未知成分が含まれている。分離カラムに最初の検体が注入された後、充填剤には未知成分の一部が吸着したままになっている。その吸着した未知成分が、分離カラム内の固定相・移動相間の分配係数を刻々と変化させる。2回目の検体を注入時には、当然のことながら、分配係数が初回の注入時とは異なってくる。計測後、クロマトグラム上で信号が出なくなり、安定した状態と見えても、カラムからは充填剤に吸着した成分が溶出している。それにもかかわらず設定した条件では検出されていない、あるいは非常にブロードな信号となり見かけ上検出さていないだけの場合がほとんどである。
3. ― 液クロが“生き物”に例えられるわけ -
3.1. HPLCカラム内で起こっている分離現象
HPLCは、溶液(移動相)が充填剤(固定相)の間を流れている環境下で、溶液中の各成分が移動相と固定相間で分配、吸着・脱離(吸脱着)する現象によって分離・精製する技法である。
この分配・分離現象をモデル的に図-1及び図-2に示す。ここで固定相、溶質及び移動相はそれぞれ下記の様に表現されている。
a)逆相系カラムを構成する代表的な充填剤であるODS(Octadecyl Silicate:(Si―O―C18H37が
「固定相」となり、その空隙に移動相の溶液が入っている「相;phase」の状態を示す(図-1、図-2)。
b)分離される2種類の溶質成分をそれぞれ「☆」及び「★」マークで示してある。
c)分配・分離現象;左側→右側へ図が時間の経過に伴い成分:溶質(「☆」、「★」)が移動相・
固定相間で分配・分離されて行く状態を示す。
したがってカラム内の環境は時間と伴に刻々と変化することからHPLCカラムは“生き物”に例えられる。
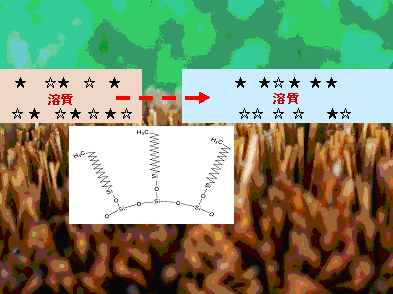
図-1 固定相:ODS、溶質及び移動相の構造的概念図
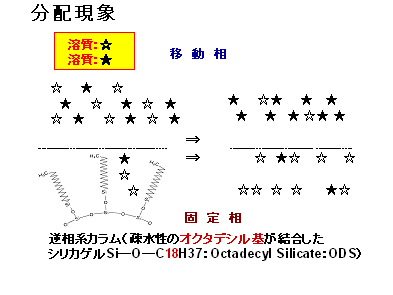
図-2 分析カラム内の成分の分配現象のモデル
充填剤の粒子サイズ;5~10μについて、HPLCで分離・精製される成分(分子)サイズとの比較概念図を図-3に示した。すなわち充填剤の粒子サイズは、溶質分子及び固定相;ODS分子サイズと比較すると極めて大きいので、分配・吸脱着現象は移動相・固定相間での分子の相互作用により進行していることを分離現象の概念として頭に入れておかなければならない。(サイズ排除クロマトグラフィーでは、分子ふるい作用ゆえに充填剤の粒子孔サイズと分離・精製される成分(分子)サイズは同水準にある)
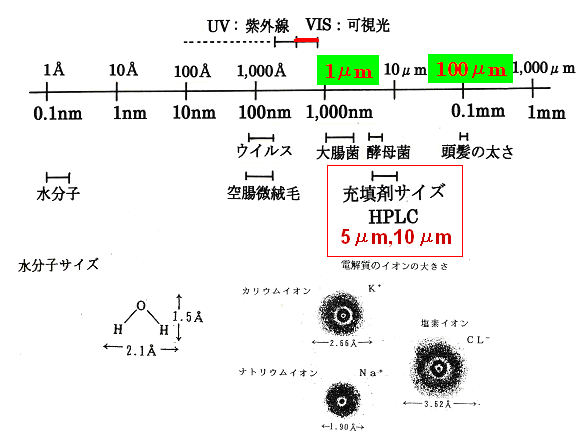
図-3 HPLCで分離・精製される成分及び充填剤サイズの比較
3.2.溶媒組成が信号の波形に及ぼす影響
標準溶液と試料溶液との成分・組成の相違がクロマトグラムへ及ぼす影響を明らかにすることを目的として現実的なモデルを想定した。
a) α-トコフェロールは化学構造としてOH基を含む脂溶性の物質である(図-4)。
これを定量するためにシリカゲル系カラム及びn-ヘキサン‐エタノール系移動相
(成分組成;n-ヘキサン:エタノール=9:1)で定量する系を設定した。
したがってα-トコフェロールは、移動相を構成するn-ヘキサン及びエタノールの両溶媒に溶ける。
b) 移動相を構成する2種類の成分であるエタノール(図-5左側)又はn-ヘキサン(図-5右側)単独で
調整したα-トコフェロールの標準溶液のクロマトグラムを図-5に示した。
c) この時のカラム内における溶質の移動の状態を模式的に図-5に示した。
d) 溶質が試料溶液の溶媒及び移動相溶液の両者に良く溶ける場合、溶質は「移動相」と伴に
移動するために正常な分離現象を示す。(図-5右側;α-トコフェロールをn-ヘキサン溶媒で調整)。
e) 溶質が移動相溶液に比べ試料溶液の溶媒に良く溶ける場合、溶質は「移動相」に依存せず
「試料溶液」の流れと伴に移動し、信号波形はブロードとなる。
(図-5左側;α-トコフェロールをエタノール溶媒で調整)。
[解説1]
・溶質(測定対象成分)の化学構造及びその溶媒特性
脂溶性ビタミンのビタミンEの化合物名は、α-トコフェロール(α-tocopherol)である。したがって化学構造
では「OH 基」を含みエタノールに可溶である。また「- CH2-」の長鎖で構成(図-4)されているためヘキサン
にも溶ける。
・測定成分(α-トコフェロール)に対する移動相を構成する各溶媒に対する溶解性
α-トコフェロールは、溶媒のヘキサンに比べてエタノールによく溶ける。
エタノール > へキサン
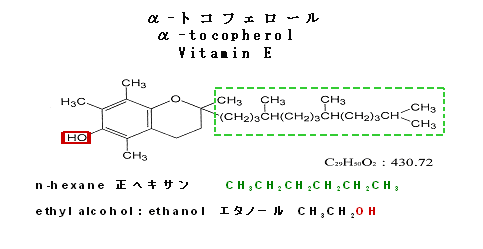
図-4 α-トコフェロールの化学構造及び溶媒特性
[解説2] 典型的な現象を示すためのモデル実験;
< HPLCによるα-トコフェロールの分析事例 >
α-トコフェロールは、ヘキサン及びエタノールに可溶である。しかしα-トコフェロールはヘキサン
よりエタノールに良く溶ける。したがって試料溶液中のα-トコフェロールはヘキサンを主成分とする
移動相系で吸脱着・分配現象により分離・精製されることなく一時的に試料溶液中のエタノールと
共に移動することになりブロードな波形となる(図-5・左側)。
他方、ヘキサン溶液では、α-トコフェロールは移動相と供にカラム内で吸脱着・分配現象の基に
展開され正常なクロマトグラムを示す(図-5・右側)。
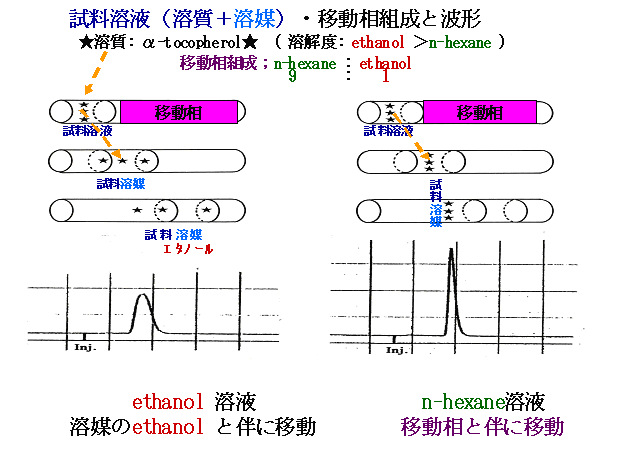
図-5 カラム内での分離現象の概念及びそのクロマトグラム
溶質(測定対象成分)が、移動相を構成する各成分に可溶であり、またカラムへ注入する溶液量が移動相の流量に対して少量であっても、標準溶液と試料溶液間で溶媒組成が両者で異なれば、当然、クロマトカラム内の環境は両者で異なり、クロマトグラムの両信号の波形が異なることが想定される。
検量線法による比較定量法では、標準溶液と試料溶液から得られる信号の強度、特に信号ピークの高さの比較による定量法であるため、両信号波形は相似形であることを前提としているので注意が必要である。
標準溶液と試料溶液の成分組成は本質的に異なることから、信頼性ある定量分析値を得るには、マトリックスの差による影響が常に伴うことを認識し、その影響が定量精度として許容される範囲にあるか否かを検証する分析法バイデーションが必要となる。
[topへ]
第8回(最終回) バリデーションの要件8) 回収率 : Recovery
(2009/11/24)
1.回収率:Recoveryとは
既知の測定対象物質を試料に添加して測定した時、計算された添加量に対する添加量として測定された定量値(量)の割合
<回収率の標準的な算出方法の事例>

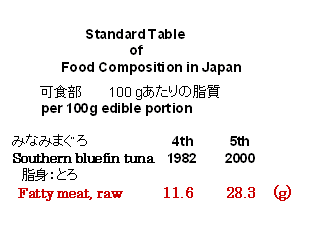
2.食品標準成分表に見られるマグロの脂質含有量
日本食品標準成分表に記載されているマグロの脂質含量について、1982年の四訂版(11.6g/100g)と2000年の五訂版(28.3/g)とを比較するとその数値は大きく異なっている(表-1)。
この数値の差は、マグロの捕獲地の違いでもなく、また養殖などによる餌の違いによるものでもない。両数値とも天然の「みなみまぐろ」の“脂身;とろ“のもので、その差は本質な魚の違いによるものではない。
これは、人の健康への影響を考慮して、人が魚から摂取する実質の脂質の量を反映させるために脂質を定量するための抽出溶媒の種類が変更されたためである。

表-1 日本食品標準成分表に記載されているマグロの脂質含量
3. 「規格と試験方法は一体」
一般に検査・分析を始める際に使用される公定書などに記載されている試験法は検査・分析方法の指針を示すもので、そのまま試験・分析に適用されることは少ない。
これら規定された試験法は汎用性のある方法で記述はされてはいるが、具体的な試料調製、操作の段階では試料の種類ごとにマトリックスの影響を考慮し試験方法を設定しなければならない。したがって信頼性ある測定値を得るためには、「分析法バリデーション」の実施が必須となり、SOP (Standard Operating Procedure); 標準操作手順書 が必要となる。したがって規格試験では、規格と試験方法が一体として扱われる。
4.添加回収率の目安
定量分析で得られた定量値は、試料溶液を構成する成分及び組成(マトリックス)の影響を受ける。そのためにマトリックスの影響を確認するために添加回収実験が必須となる。
回収率は、測定対象成分・濃度、測定物質の種類・形態、分析法などにより当然異なり一律に設定されるものではないが、その目安となるものがあると好都合である。
GC/MASSによる農薬分析に対する妥当性評価ガイドラインの事例を表-2に示す。
表-2 各濃度の水準に対する測定回数、真度(回収率)及び精度の目安

デジタル化された数値は歩きだし、やがて走り出す!
その数値の根拠は?
その数値の計測・算出の条件は?
その数値の有効数字は?
[topへ]
|