|
|
トップ>講師コラム・取材記事 一覧>
GMPの常識・非常識:情報機構 講師コラム:情報機構:情報機構 講師コラム
講師コラム:中村 宥治 先生
GMPに詳しい中村宥治先生のコラムをお届けしております。
『 GMPの常識・非常識 』
[1] [2] [3] [4] [5] [6] [7] [8] [9] [10]
第1回 ネーミングの大切さ (2007/07/02)
GMPを日本語でいうと「医薬品の製造管理及び品質管理基準」1)となる。
最近食品についても「適正製造規範(GMP)ガイドライン」が示されている。2)
[Good Manufacturing Practice]を素直に日本語に訳してみるとこの方が適切と思われる。
さて、GMPが世の中で注目され始めたのは1960年代であり、当初その意味がわからず、Gはゴルフ、Mはマージャン、Pはパチンコとネーミングし、GMPの理解に努めたものである。これを最近のことばに置き換えると、Gはゲームセンター、Mはマンガ、Pはパソコンとなるのではないか。
[Manufacturing]を英語で分解すると[Production]&[Quality control]となる3)。
[Production]&[Quality control]を日本語に訳してみると製造管理&品質管理となる。こうなると[Manufacturing]と[Production]は同じ日本語になってしまう。
日本語は難しいが、製造部門[Production division]と品質部門[Quality Unit]は互いに独立していることがGMPの基本要件の一つとなっていることを忘れてならないであろう。
医薬品GMPは、薬事法の改正により、外国製造業者にも適用されるようになるが、本来外国語であるGMPを日本語にし、更にこれを英訳し、これが日本のGMPであると紹介すると外国において日本のGMPを実施する場合、時として混乱が生じことがある。
それを避けるためには、[Manufacturing]を正しく理解することが必要である。Mは「マンガ」だけでなく、M「真心」が大切である。
1) 医薬品及び医薬部外品の製造管理及び品質管理の基準に関する省令
(H16.12.24 厚労省令第179号)
2) 健康食品ガイドライン(H17.2.1 食安発第0201003号)
3) EUGMP:EC委員会指令
[topへ]
第2回 四文字熟語 (2007/07/17)
一般的な話として,四文字熟語は、どちらかというとアナログ語である。何となく理解出来るがほんとのところは判らない。
薬事法1)の中で「品質管理」という四文字熟語が数多く使われているが明確に定義されていない。従って、世の中で一般的にいわれている「品質管理」に相当し、英訳すると「Quality control」となる2)。
例えば、GQP省令と呼称されている「医薬品、医薬部外品、化粧品及び医療機器の品質管理の基準に関する省令」3)として「品質管理」という四文字熟語が使用されているがこれを英訳すると「Quality Assurance」4)となっている。
一方、原薬GMPガイドライン5)においては、「品質管理」は、規格に適合していることを確認又は試験することをいうと定義されていて、英語では、「Quality control」となっている。
「品質管理」ということばを正確に伝えようとした場合、「品質管理(QA)」又は「品質管理(QC)」と表記するとよいのではないか。
「品質管理業務(Quality Assurance duty)3)」とは、医薬品等の市場への出荷の管理、製造業者及び外国製造業者に対する管理監督、品質情報及び品質不良の処理、市場からの製品の回収処理と規定されている。
「品質管理(QA)」の業務は、日本語を正しく理解し、「患者」さんに対し、自らの製造(Manufacturing)行為についてつつみ隠さず説明出来ることが肝要である。
1) 薬事法(昭和35年8月10日 法律第145号)
2) ISO9000(2005)
3) GQP省令(H16.9.22 厚労省令第136号)
4) GQP省令の仮英訳について(H17.9.9 事務連絡)
5) 原薬GMPガイドライン(H13.11.2 医薬発第1200号)
[topへ]
第3回 カタカナと漢字の日本語 (2007/07/31)
日本語には、平仮名、カタカナ、漢字、英文字の略語などいろいろな表記法があり、世界で最も情緒的な言語といわれている。
例えば、生物薬品では人とヒトは意味が違うようである1)。同じように、医薬品GMP基準2)でいう「バリデーション」とISO9000でいう「妥当性の確認(Validation)」は必ずしも同意語ではない。
医薬品に限らず多くの工業製品は,開発・設計~試作・試製~生産・流通という「過程」を経て世の中で販売することが出来る。
シュハート3)は、この「過程」を図に示すように説明している。
即ち、1)条件検討の段階で、必要な要因を検討した後、2)鉢の状態の段階でこれらの要因をいつも同じように管理出来る状態にする(すり鉢で胡麻をすり出来上がり状態を確認する)。この状態を造り出すことで、3)将来の予測が可能となる(いつも同じ胡麻味噌ができるようになる)。
工程 → 1)条件検討 → 2)鉢の状態 → 3)将来の予測
図 シュハートの考え方(日科技連:新編統計的手法演習)
*ISO9000
1)適格性確認プロセス(Qualification process)
2)妥当性確認(Validation)
3)検証(Verification)
|
この考え方をISO9000の用語に当てはめてみると1)条件検討が適格性確認プロセス(QP)、2)鉢の状態が妥当性の確認(Validation)、3)将来の予測が検証(Verification)に相当する。
やはり、医薬品のバリデーションは、製造販売承認申請書の作成時点から、将来の予測が出来るような実施計画書(プロトコル)を作成する必要があり、そのためには、ISO9000で定義されている前提となる「適格性確認プロセス」を明確にし、検討することが最も大切といえるであろう。
1) JP 参考情報 日局通則40等に規定する動物由来医薬品起源としての動物に求められる要件
2) H16.12.24 厚労省令第136号 GMPの基準に関する省令
3) JIS Z 9021:1998(W.A.Shewhartの管理図)
[topへ]
第4回 品質情報の発生を防止するための奥の手 (2007/08/14)
薬事法上では、品質情報1)と品質不良2)は区別して考えて対応した方が良いようである。
このためには、「基準」に対する考え方について、研究部門~生産部門~営業部門を通して情報を共有化し、全社的に徹底する必要があるであろう。
医薬品の場合、法律上約束している項目と自主的に設定し自らこれを遵守している部分がある。
目標品質基準1)をどの様に設定するかは、企業としての実績に基づく積み上げが前提となる。製造や品質の管理を疑わせるような製品を出荷した場合、
「回収」を決定する基準となるものである。
薬事法では、回収の決定は、製造販売業者がなすべきとされているが、製品の特性に関する種々の技術的な諸問題を無視して回収の可否を決定すべきではない。
最近の事例2)として、製造販売業者が実施した抗生物質注射剤の破ビンによる回収手続きが、製造業者との間で波紋3)を呼んでいるが、破ビンの問題は、以前から指摘3)されていることで、取決め事項の中で、工程管理として破ビン発生時の取り除く範囲を明確にし、それについて「妥当性の確認」を実施しておくべきであろう。同時に、CAPAの観点から、モニタリング方法を検討しておく必要がある。
また、安全性の面からも、微細なガラス片が本当に混入していないのか検証する必要がある。
そこまで検討して、消費者基準であるZERO リスクに対応することが肝要である.その結果、製造販売業者として説明が出来なければ、市場にある全製品を回収することは当然の措置である。
いうまでもなく、GMPは単なるシンボルマークではない。生産部門だけでなく研究部門~販売部門まで製品の品質に自信を持つことが大切であり、特に営業部門における「品質」に関する日頃の教育と情報の共有化が品質情報の発生を防止するための奥の手となるであろう。
スカイラーク社のリスクマネジメントの方針を参考までに以下に示す。
リスク安全への合意
1)逃げるな、かくすな、嘘つくな。
2)データに基づき説明する。
3)説明内容はわかりやすくする。
4)許容レベルは安全への合意をとる。
5)基準は、購入先と自社は同じレベルとする。
6)怖がりすぎるのは易しいが、正当に怖がるのはむずかしい。(寺田寅彦)
|
1) ISO9000 3.2.5 品質目標(Quality objective)(2005)
2) じほう社 Mail news (2007.7.24)
「パンスポリンの回収で委受託間に見解の相違」
3) 今井澄栄 GMP・バリデーションの具体的実施例と査察への対応策
(1997)技術情報協会
[topへ]
第5回 無菌管理の神髄 (2007/08/28)
医薬品の製造工程は、製品の品質を確実に保証するため、有効成分を作ることを目的とする「原薬製造工程」と必要な添加物を加えて投与形態とするための「製剤製造工程」に大別されている。
同時に使用する主要な原料により、「化学薬品」と「生物薬品」区分される。
また、投与経路の違いにより、「無菌管理」が必要な場合とある一定レベル以下で微生物等の汚染を管理すれば良い場合に区分される。
更に、投与方法によっては、医療機器との整合性を必要とする場合もある。
このように、医薬品の生産には、多くの製造工程が存在するが、「無菌管理」は、その中で最も厳格な管理が必要な製造工程といえよう。
「無菌(Sterile)」とは、成育可能な微生物が存在しないことをいう1)と定義されている。無菌試験については、JP15の一般試験法で規定されているが、抜き取り破壊検査であり、無菌の定義を試験検査で実証することは現実的に困難である。
特に静脈に直接投与する場合、注射剤の中に成育可能な微生物が1個でも存在すれば、血液中で増殖し、敗血症になり、発見が遅れれば短期間で死亡することとなる2)。
最近食品のGMPガイドラインが取りざたされているが、具体的にどこまでGMPを実施するのかを考える場合、無菌のGMPの考え方を参考とすると管理レベルや管理のポイントが見えてくることがある。
例えば、作業室の環境(差圧・換気)、原材料の搬入手順、作業者の衛生管理基準、試験機器の洗浄後の確認、機器の設置方法、床排水及びモニタリング方法等々である。
その意味で、非無菌しか製造していないから、無菌管理に関心を持たないという考え方は、GMPの観点から視野を狭くし、不幸なことであるといえよう。
このような立場から考えると、パラメトリックリーリス3)が可能となるためには、前提条件として「適格性の確認プロセス」を最新の技術レベルにより、評価・検討し、実生産設備で「妥当性の確認」及び「検証」を行い、その結果に基づいた工程管理としての「検証」を継続的に実施して行くというより厳格なGMP管理が不可欠となる。
しかしながら、最新の設備を導入し、環境の連続モニタリングが可能な設備を導入したとしても、工場全体がはじめに決定した手順を正確に守り、安易な方向に流されることなく、厳しい自己抑制のもと生産管理や品質管理の業務に向き合うのだという感性が必要である。このことが無菌管理の神髄であるといえる。
無菌製造所の監査を行った経験から、作業手順について安易な妥協を繰り返し、自分たちの都合で作業のやりやすい方向に手順を変更すること認めている製造所は、データを見せられてもほんまもんの無菌製品が出来ているか疑問に思うことがある。
ほんまもんの製品のみが患者の健康被害に対するリスクを減少することが出来る。
1) 無菌操作法による無菌医薬品の製造に関する指針
2) 2006.7.17 じほうメールニュース
3) JP15 参考情報「最終滅菌医薬品の無菌性保証」
[topへ]
第6回 バイオ医薬品における汚染防止 (2007/09/11)
ドナーとは、細胞組織医薬品、細胞組織医療機器の原材料となる細胞又は組織を提供するヒトと定義されている1)。
GMPの基本的な考え方の一つとして、「バイオバーデン」2)の考え方がある。バイオバーデンは対象となる「もの」の中に存在する恐れのある微生物のレベルとタイプ(例えば特定微生物又は非特定微生物)について、ある一定のレベルを超えない又は規定した特定微生物を検出しない限り、汚染とは考えないことを意味している3)。
無菌の原材料を製造するためには、「バイオバーデン」の概念を導入することが不可欠である。
一方においてドナーであるヒトは、雑種であり、微生物を含めウイルス等多くの汚染物質を含んだ「バイオバーデン」が高い原材料(リソース)であるといえよう。
このような原材料の使用に際しては、その適格性を確認するため、あらかじめスクリーニングテストが必要と考えられているが、そのテストをすり抜け(ウインドピリオド4))、投与後、使用者に問題が発生することを覚悟し、あらかじめその対策を立て、製品を造る必要がある。
ここまでくるとバイオ医薬品GMPにおいては、単に製造行為中における不適切な取り扱いを防止するだけでなく、細胞・組織の採取から製品の使用時の医療上の必要な対応まで一貫した品質方針が必要となる。
また、ドナーからの原材料の採取及び使用者への投与は通常病院で行われるため、製造場所を出来るだけ医療現場の近くに設置し、品質の評価が容易に実施可能な体制を取る必要がある。
ある種のいかがわしさが感じられるバイオ医薬品のGMPの実施に際しては、もう一度ギルドの誓い5)に戻り、時代の流れ、医療の進歩、社会の変化に対応した倫理の基本について考えてみる必要があるであろう。
ギルドに誓い(今裕訳)
1)師とその家族に感謝し、学問と技術を濫用してはならない。
2)あらゆる措置は、もっぱら病人に役立つかどうかの観点から判断し、それに合致しなければならない。
3)生存権は、胎児をも含め絶対的に保護し、患者の希望による安楽死を拒否する。
4)医師の私生活と医療技術に関しては、清廉潔白である。
5)治療に当たっては、自分の学んだことの限るべきである。
|
医療は、生命の尊重と個人の尊厳の保持を旨とし、医師、歯科医師、薬剤師、看護士その他医療の担い手と医療を受ける者との信頼関係に基づき、医療を受ける者の心身の状況に応じて行われるとともに、その内容は、単に治療のみならず、疾病の予防のための措置及びリハリビテーションを含む良質かつ適切なものでなければならない6)。
バイオ医薬品のGMPでは、特にこのような視点が大切となるであろう。
1)医薬品GMP省令第2条第9項、医療機器QMS省令第2条第13項
2)日局参考情報「最終滅菌法及び滅菌指標体」
3)ICHQ7a 用語の定義
4)ICHQ7a 用語の定義
5)Hippocrates , B.C. 460~377 ギルドの誓い
6)医療法第1条の2(医療提供の理念)
[topへ]
第7回 衛生管理どこまで綺麗にすればよいのか。 (2007/09/25)
医薬品の製造に際して、衛生管理を行うためには、まず綺麗好きな人間となることを心がける必要がある。
GMPの三原則の一つに「Keep it clean(清潔が第一)」があるがその心は、次の様なことを意図している。
・「頭にさわりたる手を未だ洗わずして、食の器および食物に手をふれることなかれ」(道元禅師)
・きれい好きな人は異質なものに敏感になる。
・Cleanliness is next to godliness (清潔は敬神に次ぐ美徳)
人と物の動きの中で、接触による汚染の機会を減らすことが基本原則となる。
まず、医薬品に接する表面(Drug contact surface:DCS)を確定する。
一般的に、DCSは医薬品を生産する際に直接接する部分又は表面上に結露が生じる部分が該当する。
図クリーンベンチにおける無菌作業を示しているがこの場合、作業開始前に
1)DCSは、良好で、洗浄され、消毒されていることを確認するために、一定の明るさの下で、目視検査を実施する。
2)洗浄及び消毒の作業の都度、機械器具について、清浄であることの検証(Verification)を実施する。
3)使用前に手袋及び保護具の破損・清浄性を確認する。
4)消毒液は使用前に濃度及び調製法について確認する。
これらの前提条件(適格性の確認プロセス)が確認された後,無菌作業を実施する。

無菌作業中は、クリーンベンチ内のDCSが設定通り維持されていることをモニタリングする。
更に作業終了後直ちにDCSの拭き取り検査を実施し逸脱していなかったことを検証(Verification)する。
洗浄におけるCAPA(是正措置、予防措置)としては、
1)容易に洗浄出来ない部分は、作業終了後速やかに洗浄しなければならない。
2)洗浄・消毒が不十分なDCS及び手袋や保護具は作業を開始する前に是正しなければならない。作業終了後微生物に汚染されていないかを検証しなければならない。
結局どこまで綺麗にするのかということの答は、決められた手順通りに,実施し、丁寧にデータをとり、説明責任を果たすことである。
このためには、設備の維持管理が重要なファクターであることはいうまでもない。
[topへ]
第8回 自己点検の目的 (2007/10/09)
OECD1)によると、Auditは、ある組織の活動に付加価値を与え、改善することを目的とした独立の達成目標保証活動(objective assurance activity)である。
Auditは、「体系的かつ統制のとれたアプローチを取り入れ、リスク管理や統治プロセスの有効性を評価(assess)し、改善することにより、組織目標の達成を支援するとし、内部監査(Internal Auditing)は、幹部に対して報告を行う部門によって実施される内部統制(Internal Controls)のアセスメントが行われるのに対して、外部監査(External Auditing)は独立の組織によって実施される」としている。
ISOの監査(Audit)2)においても、内部監査と外部監査に区別され、内部監査は、第一者監査とも呼ばれている。GMPにおける自己点検は、内部監査に相当するものであるといえる。
監査を実施する人は、品質保証(QA)の立場で、十分な経験と知識を有する者が担当することが望ましい。
医薬品の場合、事業者は、GMPにおける自己点検の結果に基づき、改善が必要な場合は、必要な措置を採ることが法的に求められている。3)
改善が必要な場合とは、患者さんに健康被害を生じるか否かで判断すべきものである。例えば、無菌製品の製造設備が30年間変更なく、無菌製品を造り続けることは、無菌を保証する上で危険なことである。
無菌の保証方法は、「カレント」でなければならない。無菌管理のデータの精度を落とせば見かけ上無菌になるが、事故が起これば、製造において製品が汚染されたという過失に対して責任を問われることになる。
この場合、所要の措置とは、自社の設備で生産することをやめ、最新の設備を持つ他の製造業者に当該品目の生産を委託することを決断することである。
コスト面で利益が出ないのであれば、生産をやめることもひとつの選択肢である。
自己点検の目的は、GMPの現状を外部に対して情報を公開するとともに、「品質方針」に対する問題点のスピークアップ(内部牽制)を意図したものである。
このような目的を外れた自己点検は、法の定めにより定期的に実施しても意味のないものとなる。
事業者がGMP自己点検を経営の中で真剣に生かすことが「品質」に対するリスク管理に対応したことになるといえる。
1) Glossary of Key Terms in Evaluation and Result-Based Management
2) 監査(Audit)ISO9000 3.9.1(2005)
3) GMP省令第18条第2項
[topへ]
第9回 歴史は繰り返す (2007/10/23)
医・薬には永い歴史がある。
「傷寒論」1)の本文には、病気の症状、経過を述べ、それに対する治療法(漢方)をあげているだけで、特別の理屈で説明していない。
その臨床的観察が正しく、適用した漢方が有効であるなら、後世の人がそれを追試して同じ効果をあげ得る筈である。
事実を正しく把握していたら、二千年を経ても、その事実には変わりはない筈であると述べられている。
壮大な臨床研究の結果、偽物が淘汰され結果として現在の処方が認められるようになったといわれている。
一方、21世紀はウイルスのルネサンスの時代といわれている2)。
ウイルスは生物の生きる仕組みを知り尽くしている。
人間が増えすぎて生態系を破壊すると人を殺そうとし、それで人口が減れば、今度は共存の体制となる。
人は、昔は自然と接することにより、病原体を病気にならない程度に吸収して免疫状態を保っていた。
人間の復活を図るためにも、一度自然に戻る生活をしなければいけないと思う。
一方、医薬品GMPは法律として存在し、その枠組みを如何に遵守するかに目を奪われがちであるが、本来どの様な考え方で、どの様な仕組みで患者に役に立つ医薬品を検討し、製造するのかということは製造業者自身が決めなければならないことである。
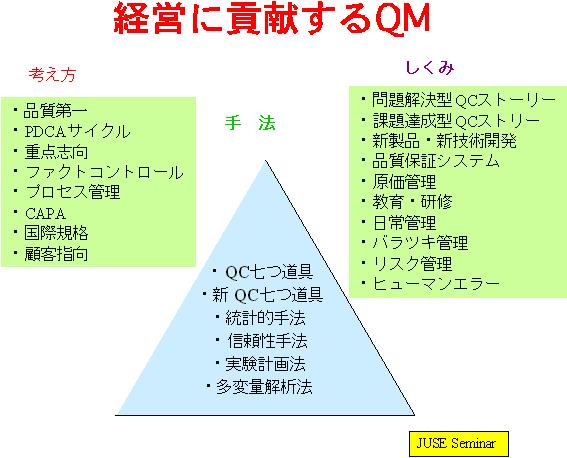
このための手法として、例えば、統計的手法、信頼性手法、実験計画法、多変量解析法、品質機能展開等の基礎的な素養を身につけることが効率的なGMPを実証する必要条件となるであろう。
下図に「経営に貢献する品質マネジメント(QM)」の考え方を示している。
バリデーション、リスクマネジメント、PATなどを論議する前に,いつか来た道を振り返って、このような素養を身につけることが大切である。今後も我が 国における世界に通用する産業の一つとしてGMPを実施する上で、このような基礎的素養を身につけることを「品質経営(Quality Management)」の課題として取り上げるべきであろう。
1) 「臨床応用傷寒論解説」大塚敬節(東洋医学選書)
2)シンポジウム「地球環境と人の暮らし」根路銘(ねろめ)国昭(2006)
[topへ]
第10回(最終回) 偽薬を見分ける (2007/11/06)
製造販売業者は、患者に健康被害を及ぼすような事象が含まれている医薬品を市場に出荷した場合、それを回収(Recall)する責任を有している。
日本は薬事法で製造、販売、流通が厳しく管理、監視されていることから、「偽薬」についてあまり関心がないが、世界医薬品市場約60兆円の約5~10%を「偽薬」が侵食しているといわれる1)。
ちなみに、EUとの相互認証に関する回収報告書様式2)には、偽物か Counterfeit/Fraud {YES・NO} を記載する項が含まれている。
GMPにおいては、如何にして作業者による間違いをなくすかに苦心しているが、出来上がった「商品」が本物であることをどのようにして証明するのかはどちらかというとこれまであまり関心がなかった。
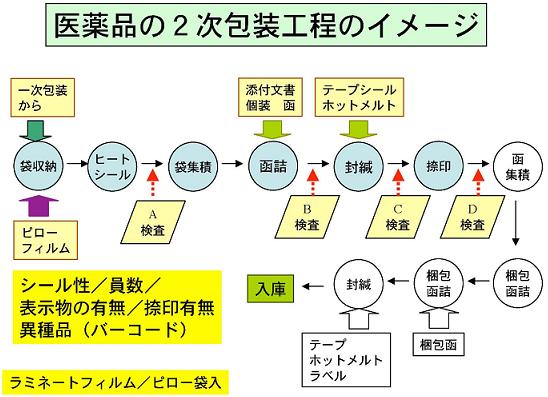
「偽薬」の対策としては、工場から出荷された製品が患者さんの体に入るまで間違いなく「本物」と「偽薬」が識別出来るようにすることが前提となる。
医療機関において使用する前に「本物」と「偽薬」が識別出来るようにするためには、図「医薬品の包装工程のイメージ」に示しているように内容物の確認より、包装材料が「本物」か「偽薬」かが識別出来るようにすることが考えられる。
当然のことであるが、製造工程で使用した材料例えば、ラミネートフィルム/ピロー包装の素材は、購入時の型番、業者名等の情報が解っている筈である。
この情報をIT-TAG により、使用前に医療機関で一致性を確認することが必要となるであろう。
その他欧米では、一部実施されている紙幣並みの偽物防止策の検討が必要となるであろう。これらの措置は現状を注意して観察しながら進めることが必要となるであろう。
CSR(企業の社会的責任)の大切さは、「いのち」に係る企業として、企業倫理の確立と実践に絶え間ない取り組みの土台となるものである3)。
このためには、良いも悪いも、情報を公開し、ステークホルダーに心を配りながら品質経営を実現する。
特に、製品安全の責任、事業活動の環境への負荷、情報管理の考え方、従業員の安全等その企業を多面的に理解して頂く上で必要な情報の開示が大切である。「病の向こうに人がいる」ことを考えれば偽物は徹底的に排除しなければならない。
1) じほう Mail news 「偽薬とTAG」2006.4.20
2) 緊急回収通報 Rapid Alert Notification of a Quality Defect/Recall
3) 長沢恵美子(経団連)医薬品企業向けのCSR
終わりに
最近の研究では、生態維持の仕組みとして、「モータータンパク質」と呼ばれる細胞内の物流基盤の役割に関心が集まっているとのことですが、GMPにおいて「品質マネジメント」は「モータータンパク質」の役割に相当すると思われます。
患者さんの役に立つ医薬品を研究開発・生産・販売するため、企業の枠を超えた品質保証部員(モータータンパク質)としての活躍を祈念しております。
長い間「GMPの常識・非常識」をお読みいただき有り難うございました。
中村先生のコラムは今回を持ちまして終了いたしました。
ご愛読ありがとうございました。
[topへ]
|
中村宥治先生のご紹介
某大手製薬企業で、工場建設や品質保証業務に携わり、その間、日薬連GMP委員 会委員を経験。
現在、NPOQA支援センターに所属し、GQP・GMPのコンサルテーション、日科技連 「GMP」関連セミナーの講演等を行う。
最近では、ベンチャー企業のバイオ医薬品GMPにも関与している。
|
|