|
第12回「まとめ
~CSV実施ポイントのおさらいと今後の課題~」
(2022/6/7)
皆様、本コラムの掲載が遅くなり申し訳有りません。いよいよ、今回が最後のコラムとなります。これまで、お付き合い頂き有難うございます。また、これからCSV活動に従事される方にとって、少しでも一助になればと思っております。
今回は、これまでのおさらいと課題について纏めさせて頂きました。コラムの最後に、少しでもお役に立つコンテンツや当方が参加している学会活動を紹介させて頂きます。実務上、困ってしまったことや、相談してみたいことなどありましたら、お気軽にご参照・連絡下さい。
■CSV実施ポイントのおさらい
コンピュータ化システムバリデーションの流れに沿って、これまでのコラムとポイントを纏め直してみました。CSVの基礎セミナーでは、より多くの内容や深堀して解説するものなど、様々あります。しかし、押さえるべき実務上のポイントは意外と単純であり、「悩んだら、一旦規則・ガイドラインに立ち返って考える」ことです。

■今後の課題
CSV実施を行うにあたり、省令・規制・ガイドライン・世界の潮流を学びながら、業務を一人で遂行することはかなり負担が大きいものです。しかし、実際製薬/医薬品製造メーカー側でCSVを実施するための体制、過去の経験、スキル、ノウハウなどの観点から、全てを網羅することは難しく、CSV実施のための人財育成や強化が十分とは言えません。
おさらいの一番最初に挙げたポイント…企業の命題は、信頼出来る製造業務プロセスを確立し、不良品が無い医薬品を人々に届け、「人々が健康である」状態にすることを実現するために、企業の経営者はGMPやCSVに関する教育・人財確保・社内での情報共有などに投資をしなければなりません。
その上で、CSVやその周辺知識を勉強するだけでなく、以下に実務が出来るようになるか?育てるか?という点が課題です。実際に「CSV活動が出来る」ようになるためには、組織的に育成する計画と育成方法を考えることが重要です。そのため、第三者の力を借りてOJTしながら育てることも有効です。
■一緒に課題を解決するための取り組み
現在、CSVセミナーを中心に教育や、Q&A方式でのアドバイスを行っておりますが、各企業様のお悩みやご相談の本質というのは、実は共通しております。それらに対して、当方では以下のような活動を通じて、本業界での研究・実践・フィードバックを通じて、少しでもお役に立てる活動を行っておりますので、是非以下をご覧頂ければと思います。
(1) 情報機構様とのe-Learningコンテンツ開発
企業様の中でもe-Learningや教育コンテンツが用意されているかと思います。現在、新たに取り組んでいるコンテンツは「ケーススタディ」に重点を置き、実際のCSVの流れを体験出来る内容を鋭意開発中です(だいぶ遅れておりますが…)。知識だけではなく、実務と照らし合わせた時、どう考えれば良いか?身に付く工夫を検討しておりますので、正式な公開までお待ち下さい。
(2)ISPE GAMP COP 分科会活動(CSV人材教育)
ISPE(国際製薬技術協会)のGAMP COP(ommunities of Practice)という場において、業界全体や特殊領域の課題に対して、業界関係者が集まり「分科会」活動を通じて成果を発表・公開しております。当方は、その分科会の一つである「第5分科会」にて「企業におけるCSV人材育成課題の検討」を行っております。
そして、2022年7月7日(木) 10:00から「GAMP 実践セミナー2022」において、各分科会での課題検討・成果をWebinar形式で発表いたします。GAMP・CSV・DIなど、今関心が高い内容が集まっておりますので、是非ご参加下さい。
(3)CSVに関するスポットドバイス
CSVを進めていくと必ずお悩みや相談事項が出てきます。そんな時、誰に相談したら良いか?どう考えたら良いか?経験が浅いCSV担当チームや担当者はお悩みではないでしょうか?
かと言って、常時CSVコンサルタントが企業にいる訳ではないため、スポットでお悩みや課題を伺い、ピンポイントで回答・アドバイスさせて頂いております。お気軽に相談出来るよう、まずはこちらのサイトにお問い合わせ頂ければ対応させて頂きますので、要所〃でご活用下さい。
(4)いつでもどこでも基礎セミナー
お気軽にCSV基礎セミナーを受講出来るよう、オンデマンド配信やDVDを販売いたしております。こちらに関しては、当方のブログ経由でご確認下さい。尚、毎年内容をアップデートした基礎セミナーは、情報機構様にて行っております。時期が決まり次第、ご案内させて頂きます。
(5)その他・ご意見・ご感想・ご要望について
これまで約1年間講師コラムをご覧頂き有難うございました。より具体的な内容や教えて欲しいことなどあったのではないかと思います。しかし、本コラムをキッカケにCSV活動の基礎、実務ポイント、本来考えるべきことを「見直す・考え直してみる」と、宜しいのではないかと思います。
そういった点で、企業内で本コラムをディスカッションの材料にCSV活動の進め方検討・人財育成・実務への活用にお役立て下さい。また、本コラムを通じて、ご意見・要望・質問など承っております。お気軽にご連絡下さい。
それでは、ここまでご覧頂き本当に有難うございました。また、別な形でCSVについてお話を伺いながら、皆様に有益な情報をご提供出来ればと思います。
------------------------
■その他CSV関連セミナー、書籍一覧ページはこちら
第11回「文書管理の重要性と管理ポイント」(2022/4/15)
今回はCSVにおける文書管理の重要性と管理ポイントについて、その実務ポイントを皆さんと共有したいと思います。特に、CSV活動において文書及び文書管理は、(個人的に)最重要項目と言っても過言ではありません。それはなぜでしょうか?
■文書及び文書管理の重要性
まず、CSVで扱う文書は、検証業務に関するバリデーション関連文書だけではなく、システム開発に関する文書及び証跡、運用フェーズ以降の教育訓練記録や点検記録など、コンピュータ化システムバリデーションとバリデートされた状態を維持するための文書を指します。
企業によって、何をCSV文書の範囲とするかは、(紙媒体・電子媒体に関係なく)文書管理基準などで定められているかと思います。特に、次の文書をCSV担当者は良く確認しておく必要があります(企業によって名称は異なります)。
● 文書管理SOP
● 文書リスト or 文書一覧表
● 文書の記録媒体指示書
● 文書の記録保管SOP
● 記録(テストの証跡)の保管方法
● 文書の廃棄・記録SOP…等
多くの製薬/医薬品製造会社では、上記内容に従って管理するため「文書管理システム(ワークフロー機能付き)を利用していることが多いかと思います。システムを利用しない方法で管理することも可能かもしれません。
しかし実際に、いつ、だれが作成・審査・承認した正式な文書なのか?、その承認履歴や変更履歴を管理することは困難でしょう。つまり、「文書は常に発行、改訂、廃止、回収、及び改訂に係る履歴が保存すること」が文書管理の肝になります。また、これらの文書は「ライフサイクル」に従って管理される、ということも重要です。
■文書及び文書管理のポイント
● CSVにて扱う文書とは何を指すか?、その中には開発文書や記録が含まれているか?
● 保管されている文書の一覧表やリストをすぐ示すことはできるか?
● 文書の発行、改訂、承認、廃止、回収の手順は?
● 文書の承認者は誰か?
● 文書の階層はあるか?階層ごとに承認者は異なるか?
● 文書の改訂、追記の手段が定められているか?また、それらは文書化されているか?
● 文書間の一貫性や整合性は確保されているか?参照すべき上位文書は明記されているか?
● 承認日と発効日との間に猶予期間、教育訓練期間はあるか?
● 発効・改訂発効した文書の配布・回収の手順は?
● 回収した文書は、確実に回収されたことを示す記録が残されているか?
● 文書は従業員が容易に閲覧できるようになっているか?
● 印刷された文書の有効期間、回収、廃棄の手順は?
● 最新版もしくは有効な文書であることは、どのように確認できるようになっているか?…等
■文書及び署名が電子的なものである場合のポイント
● 文書は電子版が正式か?、紙が正式か?
● 文書ははすぐに入手可能な状態になっているか?
● 文書は原本と副本(複写)を保管しているか?
● 副本は(模写)は電子的か?、機械的な複写か?
● 文書に電子署名を用いる場合、電子署名ソフトウェアとしてどのようなものを使用しているか?
● 上記のソフトウェアは、電子署名が認証され、保証されているか?
● ID付与の担当者リストはあるか?(共用IDは存在しないか?)
● IDを付与・認証する責任者は誰か?
● 電子署名が悪用されないよう、電子署名をソフトウェアにおいて、IDの使用・操作履歴を記録・保存できるようになっているか?
● IDのパスワード設定及び変更頻度は、一定の基準に基づき定められているか?…等
■日本における文書管理は「紙文書」が中心だからこそ、電子化された文書は要注意!
製薬/医薬品業界における文書管理について、日本と欧米では大きな差が1点あります。それは、日本は「紙文化」という点です。欧米では、CSVに関する文書が予め電子データで作成・管理(ワークフローによる電子承認含む)されていたり、一部の工程では自動化されるなど、効率性が求められる傾向にあります。
しかし、日本での内部監査や当局による監査は、紙で確認するのが一般的です。そのため、どの文書が正しく、最新で、承認された正式版なのか?ハンコリレーや、他の紙文書との繋がりを目視確認することが中心です。私がCSV担当をしていた頃、いかに監査で指摘されないような文書及び文書構成になっているか?エビデンスと突き合わせながら、目を皿にして検証していました。
その中で、特に気を付けないといけないと感じたことは、電子化された文書の内容が本当に正しいものかどうか?ということです。例えば、IQやOQなどのテストを実施した際に、紙で印刷したテストのチェックリスト(結果入り)と、それを電子化した際に転記ミスをしているケースなど、多数の誤りを見つけてきました。これは、ほんの一例に過ぎませんが、電子化された文書には「リスク」が含まれていることに注意が必要です。
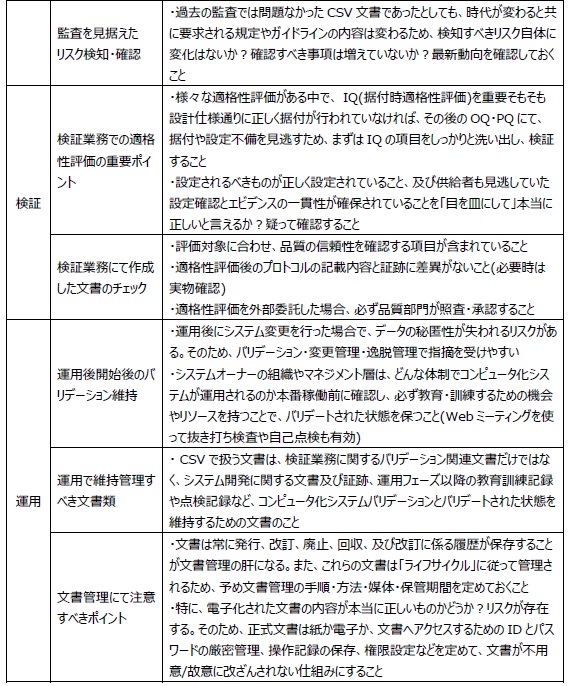
実際、「FDA警告書」に掲載されたWarning Letterの中からコンピュータ化システムの管理に関する指摘事例について、 2016年~2019年に発出された指摘事例をシステムのライフサイクル・・・開発・検証・運用の各段階に整理した内容が下図の通りです。指摘の中を見ると、文書及びデータに関する指摘が、各段階で見つかっていることが良く分かります。しかし、これは私見ですが、氷山の一角に過ぎないと考えています。
文書管理の実務において、上記のようなことが起こり得る、当局から指摘されることを踏まえ、自社の文書でも同じことが起きていないか?このような過去の指摘事例を参考に、自己点検する必要があると考えます。さらに、開発・検証段階でこれらの指摘が発生しないよう、これまでに前述したポイントをチェックしながら、リスクを「潰しこむ」ことが、CSV活動において重要な作業と言えます。
第10回「作成した文書のチェックポイント②
~監査にて問われるポイント~」(2022/4/15)
第9回のコラムから間が空いてしまいましたので、今回は第10回と第11回を連続で寄稿させて頂きます。特に、今回の2つの記事は、実際の現場における「CSVの教育とその徹底」が、いかに企業にとって大事なものであるか?重要な内容となります。
特に、CSV担当者にとって、自分一人ではなく、いかに周りを巻き込んで「バリデートされた状態を維持するか?」社内の関係者で話し合い、対策・是正していくか?がポイントになります。是非、今回のコラムを参考に社内でのCSV活動に活かして頂ければと思います。
■CSV後の文書チェックの重要性
前回は、「検証業務」における作成文書のチェックポイントを紹介いたしました。基本的に、この検証業務を通じて品質の信頼性が担保されれば、コンピュータ化システムはバリデートされたことになり、運用フェーズに入ることとなります。ここで重要な点は下図の通りです。

もし、運用後にシステム変更を行った場合で、データの秘匿性が失われるリスクがあります。そのため、バリデーション・変更管理・逸脱管理で指摘されてしまいます(中程度以上の不備指摘の内、約50%を占める※)。そこで、今回は変更管理にスポットを当てたいと思います。
※平成29年3月8日独立行政法人 医薬品医療機器総合機構「PMDAのGMP調査における 最近の指摘事例等」より抜粋
■変更管理を確認する視点
【確認すべき文書例】変更管理SOP、変更管理ログブック
● 年間の変更申請の件数は何件あるか?、(必要時)ログブックを提示できるようになっているか?
● 変更はその重要度等で分類するか?、その分類の基準はどのように規定されているか?
● 変更申請はどの部門で受領されるか?
● 変更は受領時に審査するか?、変更申請を受領しないケースはあるか?(あったか?)
● 変更申請はどのように審査(評価)されているか?
● 変更に必要な試験・バリデーションはどの部門が決めているか?
● 変更の評価の段階で不適合が見つかれば、変更前に戻すのか?
● 変更の影響調査は、どの範囲まで検証するか?
● 変更の影響調査は、年次照査で行っているか?
■基本的なポイント/経験則から伝えたいこと
● 変更管理の基本は”変更しないこと”である。その上で、変更が発生した場合に、そのメリットとデメリットを評価するための検証・検証方法・判断基準を定めているか?確認した上で、それらが文書化されていること。
● 変更管理がどの時点で発生したか、その変更が変更管理SOPに則って変更申請が、正しく・漏れなくなされているか、その証跡を確認すること。
● 申請された変更は、変更の及ぼす影響、変更リスク及び潜在的なリスク、変更の要否、変更に対する対策、対策の妥当性がCSV担当者を含め、関係者間でレビューされていること。
● 上記対策について、システムのバグや不具合だけなく、そのシステムにより作成されたデータの誤りや操作手順書との相違など、「逸脱」が認めらた場には、必ず逸脱管理が行われていることを確認し、文書として変更管理の申請と関連付けられていることを確認すること。
● 運用フェーズ直後、あるいは変更が頻発する場合、バリデーションしたコンピュータシステム又は検証業務そのものに問題がある。変更管理以前に再度検証業務した内容(テスト計画・プロトコル・証跡)と実機の再確認、及びサプライヤを交えた調査を行う必要がある。
● 変更管理の影響度は、会社の外部環境や内部環境の変化に応じて、変化するものである。そのため、影響調査自体の観点や影響度に変化が無いか?常に情報収集し、少なくとも年次単位で見直す必要がある。また、これら見直した内容に関しては、必ず教育計画を立て、教育・訓練(及びその記録)を実施し、実業務に反映されなければならない。
■運用フェーズ以降は教育・訓練が重要
コンピュータ化システムバリデーションは、システム開発におけるV字モデルに対応した検証業務として紹介されることが殆どです。これは、もちろん正しいですが、製薬/医薬品製造メーカーにとって重要なことは、バリデートされた状態をいかに保ち続けるか?そのための体制・教育・点検・業務への定着です。
実際、コンピュータ化バリデーションが対象とするのは、ソフトウェアとインフラに大別されます。ソフトウェアはバリデーション、インフラはクオリフィケーション作業を通じて、品質の信頼性を検証業務で確認います。しかし、運用後もバリデーションを維持するためにはソフトウェアベンダ、インフラベンダ、運用に関わるメンバ含めて変更管理の重要性を理解し、教育しなければなりません。
特に、運用メンバーは外部ベンダからの常駐者も多く、CSVの知識はありません。自分たちが運用しているシステムがCSVの対象かどうかも知らぬまま、運用しているケースは少なくないのです(筆者の経験では、実際に見たことがない)。そのため、変更管理がおざなりになっているケースが良くあります。
CSV担当者だけではなく、システムオーナーの組織やマネジメント層は、どんな体制でコンピュータ化システムが運用されるのか本番稼働前に確認し、必ず教育・訓練するための機会やリソースを持つことで、バリデートされた状態を保つことを理解しておかなければなりません(経営陣の関与)。
第9回「文書作成時のチェックポイント①
~監査にて問われるポイント~」(2022/2/10)
早いもので本コラムも残り4回となりました。これまである程度CSVの工程に沿って、CSV担当者が悩みやすい部分や知っておくべき点について、実例を踏まえて実践的な進め方やポイントを解説して参りました。回数も限られておりますので、以下に今後のコラム予定を記載させて頂きます。
・ 第9回 作成した文書のチェックポイント①
・ 第10回 作成した文書のチェックポイント②
・ 第11回 文書管理の重要性と管理ポイント
・ 第12回 まとめ ~CSV実施ポイントのおさらいと今後の課題~
医薬品製造販売業界において、GMPの対象となるシステムは国際基準であるGMP/QMSです。監査の最大の目的は、ガイドラインに基づき、その差異を確認しながら、CSVを実施した製造所・業務・コンピュータ化システムに潜在するリスクを評価すること。および、今後予測されるリスクを推測・評価・指摘・是正を行うことです。その中でも肝となるのが、CSV文書一式となります。
昨年から続いているGMP違反の中でも、手順書通りに作業を行っていなかった、規定に基づいて複数名で作業を行っていなかった、そしてこれらの記録(文書)が適切に作成・管理されていなかったことが、一要因として挙げられます。本業界において、紙・電子データに限らず文書そのものが正確であり、適切に管理されていることが、重要視されます。そこで、今回は「CSVに関係する文書に対して、監査時にどのような点を見られるか?」解説します。監査人の視点で最初からCSV文書を作成するアプローチは、実務的にも大変有効です。
■監査にて問われるポイント(例)
ここでは、監査においてどのような点を確認されるか?その際に必要な文書は何か?いくつか例を挙げていきます。全て記載すると、それだけで研修が出来てしまうほどなので、まずは基礎的なポイントを押さえましょう。
(1)バリデーション計画(VP)について
・ バリデーション計画書を示して下さい。
・ CSVの対象の対象範囲を示して下さい。
・ バリデーション計画書を作成・承認するのは誰ですか?
・ CSV実施時の逸脱は、逸脱報告書に記載され、バリデーション報告書に結果が記載されますか?
★ポイント
・ 前提として、企業内にコンピュータ化システムバリデーション基準(或いはそれ相当)の文書があること。
・ CSVの対象範囲が、業務に関わるシステムと周辺システムを含め、リスクベースで考えられていること。
・ 文書の作成・承認者が、予め決められていること。また、関連部門を適正に認識していること。
・ 逸脱発生時の対処法と所定の書式が規定され、正しく記録・保管すること理解していること。
(2)適格性評価(DQ/IQ/OQ/PQ)について
・ 機器装置のクオリフィケーション基準を示して下さい。
・ 評価を行う際の計画書、プロトコル、評価後の証跡を示して下さい。
・ 適格性評価(特にIQやOQ)を外部業者に委託していますか?
・ 外部委託した適正評価の結果は、品質部門にて照査・承認されていますか?
★ポイント
・ 機器装置やハードウェア類は、使用前に適格性評価が行われなければならないこと。
(ソフトウェアはバリデーション、ハードウェアはクオリフィケーションであることも合わせて理解しておくこと)
・ 各適格性の目的を理解し、CSVの対象範囲や特徴に合わせた計画書が作成されていること。
・ プロトコルは評価対象に合わせ、品質の信頼性を確認する項目が含まれていること。
・ 特に、適格性評価後のプロトコルの記載内容と証跡に差異がないこと(必要時は実物確認)。
・ 適格性評価を外部委託した場合、必ず品質部門が照査・承認することを理解していること。
■文書作成=予めリスクを挙げておくことの重要性
CSVを通じて作成された文書は、CSV担当者、業務部門、外部業者など様々な組織や担当者によって作成されます。ここで重要なことは、開発工程よりも前段階で実施する「システムリスクアセスメント」「バリデーション計画書」「サプライヤ評価」にて、どんなリスクが内在しているか?それらのリスクが文書にどのように現われるか?を先に予測し、潰しておくことが重要です。
過去の監査では問題なかったCSV文書であったとしても、時代が変わると共に要求される規定やガイドラインの内容は変わるため、検知すべきリスク自体に変化はないか?確認すべき事項は増えていないか?最新動向を確認しておくことが重要です(例えば再バリデーション)。こうしたリスクベースアプローチでの文書作成が、CSV文書作成をチェックする際に役立ちます。
第8回「コロナ禍におけるCSV推進の悩みと対応方法」(2022/1/18)
毎月本コラムをお読み頂いている読者の皆様、明けましておめでとうございます。本年もどうぞ宜しくお願いいたします。記事の公開が遅くなりましたが、新年早々新型コロナウィルスの爆発的感染により、社会や経済活動に影響が出始めており、不安な毎日を過ごされているのでは…と感じております。
実は、CSVの推進においても、コロナ禍での推進方法にも悩みが出てきているようです。これは日本だけではなく、特に爆発的な感染が発生している欧米諸国では、大きな課題の一つとなっています。今回は「サプライヤ評価」を例に、実際に起きている事象とその対応方法について、最新事例を交えて紹介したいと思います。
【事例】 現地・現物でのサプライヤ評価ができない
サプライヤ評価は、新規に取引を行う供給者(ITベンダーや装置提供者など)に対して、信頼出来る業者かどうか評価する工程です。多くの場合、書類回答+供給者側への現場訪問にて、評価対象となる文書類の確認や製造・品質の責任者などと、具体的な質疑応答を行います。
しかし、コロナ禍において企業訪問が難しくなり、本来「現物確認」が必要な文書類・証跡・会社としての物理的/情報セキュリティ・品質確保の取り組みの確認が難しくなっています。
【対応例】 スタンダードになりつつあるWebミーティングの活用と工夫
そこで、現在Webミーティングでこの点を補う/工夫し、極力供給者企業に行かずに評価する方法が試みられております。まずは、サプライヤ評価の中でも現物確認及びチェックしたい点を予め供給者に伝えておき、サプライヤ評価に必要なメンバを集めてもらいます。
その上で、Webミーティングを開催し、供給者側が外部に持ち出すことが出来ない文書や証跡などを、供給者側の担当者から説明してもらいます。文書によっては、Webミーティングの画像が荒くて見にくいがあったりしますので、その場合は画像のスナップショットを提示/表示してもらうと良いようです。
また、何度もミーティングを重ねたり、見逃しがなかったどうかを確認出来るよう、両者合意の下でWebミーティングを録画することで緊張感が増し、Webミーティング自体、サプライヤ評価における証跡にもなります。
【応用】 ”抜き打ち検査”にWebミーティングを使ってGMP違反を未然防止
日本におけるCSVの検証対象業務は開発業務ですが、昨今発生しているGMP違反の事例から、CSV完了後(運用開始後)に変更管理・逸脱管理・SOP・文書管理・情報セキュリティを指摘されることが多くあります。そのため、運用業務以降”いかにバリデーションされた状態を保っているか”を自己点検する必要があります(FDAによる指摘事項も増えております)。
しかし、実際問題として…自己点検と実務者が製造工場内で十分機能しているとは言い難い状況にあります。そこで、GMP違反が起きないようにするため、社内に設けられているCSV委員会(或いはそれ相当の部署と責任者)による製造工場の抜き打ち検査や自己点検に、Webミーティングを活用することは大変有効です。
実際の現場で使用されている文書類や記録を抜き打ちで点検し、本来の手順と異なる部分は無いか?残すべき証跡は揃っているか?工場の従業員自体、認識不足は無いか?含めて、CSV委員会のメンバーと製造工場側の主要責任者にヒアリングし、一緒に実態把握を行うことが出来ます。
【終わりに】 コロナ禍だからこそ企業として一環した品質管理と保証に注力する
工場側にある品質管理や品質保証部にとって、このようなCSV後の自己点検の重要性と具体的なチェック機能のスキルを共有でき、企業全体としてGMP順守の意義・意識・行動を浸透させることが出来ます。もちろん、ここでもWebミーティングは録画しておき、虚偽報告を生ませず、バリデーションされた状態での製造業務とシステム利用に対する理解を教育する機会にもなります。
昨今のGMP違反により経営陣の責任・品質管理・社員への教育が求められ、改訂GMP省令内でも規定されている重要な内容です。コロナ禍だからこそ、なかなか現地を確認出来ない状況を、Webミーティングを利用して積極的に確認することで、GMP・CSVの重要性理解や教育を行う良い機会になります。是非、取り入れられる部分について、自社で試行/実施されてみて下さい。
第7回「IQ(据付時適格性評価)の実務ポイントとは?」(2021/12/13)
第6回コラム「適格性評価にあたり、必要最低限必要なチェック項目(プロトコル)はどこまでか?」では、IQ(据付時適格性評価)の重要性と理由を解説いたしました。今回はその続きとして、「IQの実務ポイント」について、皆さんと一緒に学んで行きたいと思います。尚、今回紹介する実務ポイントは、筆者の経験則に基づくものであり、以下の実務ポイントが全てではないこと、条件・環境・据付物によって異なることを予めご了承下さい。
■IQにおける検証項目のおさらい
コンピュータ化システム適正管理ガイドラインに掲載されているIQ定義内容を要約すると、以下の通りとなります。またプロトコル(確認項目)に関し、ハードウェアは「機器番号」「項目名」「型式・型番」「製造者」「数量」、ソフトウェアは「インストール対象の機器番号」「項目名」「バージョン」「製造者」「数量」などを検証します。
”IQは、ハードウェア及びソフトウェアが、設計された通りの構成で、仕様書どおりの環境に正しく設置され、インストールされたことを検証する。据付時適格性評価計画書には、原則として以下事項を記載する”
1) 据付時適格性評価の対象となる文書名
2) ハードウェア構成情報及び設置場所
3) ハードウェアの温度・湿度・振動等の条件
4) 電源・接地等の設置条件
5) 通信、入出力に関する仕様
6) ハードウェアの設置の確認方法
7) ソフトウェアのインストールの確認方法
8) 据付時適格性評価における判定基準
9) スケジュール
10) 責任者及び担当者氏名
■ポイント① プロトコル作成は供給者を交えて作成する
前述の定義には含まれていませんが、実務ポイントの一つとして、計画書と合わせて「確認方法」をより具体的にした「プロトコル」のフォーマット(確認項目を含む)を、この段階で準備することが挙げられます。
この目的は、実際の確認方法が現実的であり、かつ判定出来る内容になっているか?計画書の段階でCSV責任者・担当者、及び供給者と認識合わせすることで、確認項目と確認方法の精度に誤りや認識の差異が無いことを確かめるためです。
■ポイント② 正しい検証のために正しい条件・状況を確認する
2つ目の実務ポイントは、供給者に対して「実際に検証出来る条件・環境を用意出来るか?」「異なる条件や環境だった場合どのように対応するか?」をヒアリングしながら、プロトコル作成と確認方法を固めることです。そもそも検証出来ない条件や状況をでの確認作業は、それは正しい結果ではないからです。
そのため、実環境の条件や場所が確保出来ない場合、どのように確認すれば本来の条件と同じとみなせるか?予め対応方法や判定基準を検討することが出来ます。
■ポイント③ 設定されたソフトウェアの各種パラメータと実機を確認する
3つ目の実務ポイントは、ソフトウェアが設定通りに正しくインストールされていることを確認するため、供給者(この場合はITベンダー)が用意した「設定書兼結果報告書」+「エビデンス(画面のハードコピー)」の信憑性を疑うことです。
カテゴリレベル1・3のソフトウェアは「構成設定」に変更が加えられていないため、IQを行っても不備が生じることは稀です。これに対し、カテゴリレベル4・5は「構成設定」に変更が加えられており、設定書通りにインストールした際に、ソフトウェアの仕様により変更したはずの設定内容やパラメータがデフォルト値に戻るケースがあります。
供給者側は、「設定内容に対してインストールした」ことを重視する傾向にあり、エビデンスの取得がおざなりになったり、後から異なる日時でエビデンスを取得することがあります。これらはエビデンスが持つ「帰属性」「同時性」「正確性」「完全性」「一貫性」のいずれかが欠けてしまうため、信頼性を確保するためのエビデンスとは言えなくなります。
そのため、設定ミス・エビデンス不足により、本当に設定通りにソフトウェアがインストールされていないケースが見つかったら、必ずCSV担当者は「設定書兼結果報告書」と「実機の直接確認」にて、目視での検証活動を行うことを徹底し、信頼性の確保に務めるようにしましょう。
■供給者に対するGMPやCSVの教育も必要
これまでの筆者の経験上、供給者側からGAMP・GMP・GQP・CSVに精通している人が、実際のプロジェクトに出てくることをほぼ見たことがありません。精通とまではいかないものの、過去のプロジェクトにおいて製薬/医薬品製造販売業者のGMPやGQP配下の業務プロセスに対して、ハードウェアやソフトウェアを導入し、バリデーションの経験に触れたことがある方はおります。
しかし、これはあくまでも製薬/医薬品製造販売業者からの依頼に応じて対応した経験に過ぎず、業界特有のガイドラインやルール、必要文書類、信頼性確保の重要性を100%理解している訳ではありません。また、供給者に対するRFPの中には、GMPやCSVに対する知見がある人/経験者が記載されているケースがありますが、それがどの程度ものなのか?より明確にしないと期待外れになることが多々あります。
供給者内で、これらの知識教育を定期的に行うだけの余力はないため、必要に応じて依頼側が概要説明や教育を行う必要があると、筆者自身は考えています。もちろん1回だけの教育では供給者側に知識が身に付くことは無いため、開発フェーズごとに段階的に教育する方法が一番効果的だと考えています。
ここまで行う必要性があるかどうかは、各企業や組織の判断になりますが、大前提として「契約委託者も契約受託者(供給者)もコンピュータ化システム適正管理ガイドラインが適用される」ことを覚えておくと共に、供給者に対して「CSV担当者が行う検証業務」の重要性を理解する仕組みを創ることが最大の実務ポイントでは無いかと思います。
第6回「適格性評価にあたり、必要最低限必要なチェック項目(プロトコル)はどこまでか?」(2021/11/16)
第4回コラムの続きとして、「コンピュータ化システムバリデーション(CSV)はどこまで実施すれば良いか?」について、皆さんと一緒に学んで行きたいと思います。今回は、「適格性評価にあたり、必要最低限必要なチェック項目(プロトコル)はどこまでか?」について、2回に分けて解説いたします。今回は、「重要視したい適格性評価とその理由」についてです。
■適格性評価の悩み
適格性評価…DQ・IQ・OQ・PQについて、コンピュータ化システム適正管理ガイドラインでも規定はされています。しかし、いざ実務で実施しようとすると、「何が検証されれば、信頼性が確保されたとみなせるか?」「そのためには、何をチェックすれば必要十分なのか?」悩む方が多いです。
加えて、ER/ES指針やDIに関する要件に抜け漏れがなく、それらが適格性評価のプロトコルに含まれていることを確認することも重要です。昨今のFDA Warning Letterでは、これらの不備も指摘されており、要件定義・設計・検証段階のどこかに原因があるとされています。そのため、CSV担当者は「自分が適格性評価を行うコンピュータ化システムが、どのような規制要件に対応しているのか?対応しなければならないのか?」予め確認しておく必要があるため、どうすれば良いか悩んでしまうようです。
■経験則から得たIQ(据付時適格性評価)の重要性と理由
前述した指摘漏れが生まれないように、CSV担当はどこに力点をおいて適格性評価を行えば良いのでしょうか?DQ・IQ・OQ・PQのいずれも重要ですが、私個人の見解としては「IQ(据付時適格性評価)」です。これは、カテゴリレベルと適格性評価の関係に加え、私の実経験から以下に理由を記載します。
・ IQはカテゴリレベ3以上で必要な活動であり、要求仕様及び設計仕様に基づいて、ハードウェアが据え付けられ、プログラムがインストールされていることを確認・明文化します。最終的には、PQにてURSに記載した仕様・機能を確認することになりますが、そもそも設計仕様通りに正しく据付が行われていなければ、その後のOQ・PQ(場合によっては供給者監査)にて、据付や設定不備を見逃してしまいます。
・ 供給者によるハードウェア或いはソフトウェアの据付確認及び文書に誤りがあり、かつ医薬品企業の受入テストが100%正しいとは限りません。CSV担当者は文書とエビデンスを確認しながら信頼性を評価しますが、供給者から提示された文書とエビデンスだけで評価するため、据付時適格性評価の粒度や確認項目が足りないと、不備を見逃してしまいます。
・ コンピュータ化システムの稼働日まで時間的余裕が無い場合、供給者による据付確認、医薬品企業側の受入テストの内容が、歯抜けの内容になっていることが非常に多いです。この時、システムオーナー部門の意向に基づき、稼働優先となると検証業務で指摘した内容がクリアされない(検証自体が甘い)まま稼働してしまうと、稼働後すぐに不具合が見つかり逸脱報告書の嵐となります。
■CSV担当者は信頼性確保の砦!
カテゴリレベルによっては、IQの後に行われるOQ・監査・PQの確認内容も重要ですが、まずは設定されるべきものが正しく設定されていること、及び供給者も見逃していた設定確認とエビデンスの一貫性が確保されていることを「目を皿にして」本当に正しいと言えるか?疑って確認しましょう。
私が経験した一例ですが…ソフトウェアベンダーから提供された設定内容及びエビデンスを検証した結果、こんなことがありました。
・ テスト結果を機械的に「OK」と書いているが、実際確認すると「NG」がある
・ テスト項目で期待される結果と実機の設定が異なっている
・ テスト結果の証跡となるエビデンスを取得していない/間違っている
・ 設計書に誤りがあることに気付いていないため、設定確認時に誤りを気付かない…等
そのため、多数確認し類似するミスが隠れていないか?設計書・設定時のチェックリスト・ベンダー側によるダブルチェックを依頼したことがあります。その数…二桁に昇ります。たまたま、当方がITベンダー出身ということもあり、据付時適格性評価にて細かく確認したことで、設定不備を発見することが出来ました。
しかし、CSV活動を進める方が、設備・IT機器・ソフトウェアなどに精通しているとは限りません。IQ一つ取っても、重要なポイントが沢山あるため、実務上のポイントを次回のコラムで紹介したいと思います(まだまだ伝えきれていない内容がありますが、その点はご容赦下さい)。
尚、上記のご質問やお悩みについて、2021年2月に「さらに理解が進む!応用できる!事例とQ&Aに基づくコンピュータ化システムバリデーション2021」と題したセミナーを開催しております。気になる方・会社様がおりましたら、情報機構様までお問い合わせ下さい。無料でご相談を承ります。
第5回「CSVの対象範囲はどこまで考えれば良いか?」(2021/10/8)
第4回コラムの続きとして、第5回~第7回まで「コンピュータ化システムバリデーション(CSV)はどこまで実施すれば良いか?」について、皆さんと一緒に学んで行きたいと思います。今回は、前回紹介した3種類のお悩みの内、「CSVの対象範囲はどこまで考えれば良いか?」について、最もご質問が多い内容を解説いたします。
■CSV活動で必要となる文書と悩み
良く頂くご質問かつ基本として押さえておくべき点として、「CSV活動で必要となる文書」が挙げられます。コンピュータ化システム適正管理ガイドラインでも規定はされていますが、「最初に何を準備すれば良いか?」「最終的にどんな文書が揃っていれば良いか?」という点で悩まれる方が多いようです。
これは、CSV活動を進める際、「何から実施すれば良いか?」という悩みに近く、「活動の成果物である文書」が分かれば、まずはその文書作成に必要なインプット情報の入手から活動を始めれば良いことになります。
■作成すべき文書の考え方
CSV活動を進めるにあたり、コンピュータ化システムの特性・検証範囲・作成すべき文書で悩む前に、根本的に押さえて置きたいことは、以下の2点だと考えます。まずは、「文書ありき」で捉えることで、CSV活動の内容が理解出来るようになります。
・コンピュータ化システムのライフサイクルに応じて作成する文書は何か?
(開発~検証~運用業務に関する文書)
・検証業務に必要な文章には何が必要か?(CSV活動を通じて信頼性を確認する文書)
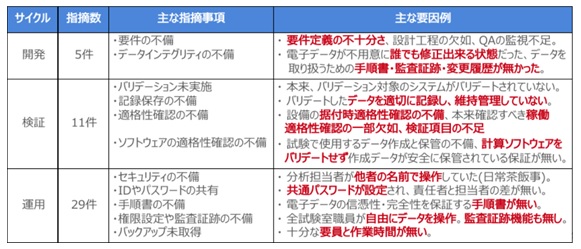
■作成すべき文書と実務上のポイント
下図に前述の2点を踏まえた「作成すべき文書(例)」の一覧を紹介いたします。画像の上のクリック先をから、全ての文書一覧が表示されますので、ご覧下さい。
CSV作成文書一覧 (こちらをクリック)
作成する文書には大きく分けて2種類存在します。1つは企業側が作成する文書、もう1つは供給者が作成する文書です。上部の例は、供給者がITベンダーの場合を想定したものとなっております。また、企業側の作成者には業務部門とCSV担当者に分かれています(厳密には、それぞれ責任者・担当者に別れます)。その上で、実務上のポイントとして2点挙げます。
1点目は、これらの文書一覧は「バリデーション報告書」の添付資料として作成することになるため、「バリデーション計画書」作成時に文書一覧を準備しておくことです。それにより、どのタイミングで、誰が、どんな文書を作成すべきか分かるようになります。
2点目は、コンピュータ化システムの開発計画・要求定義・設計・プログラミング・システムテスト・受入れテスト・運用といったライフサイクル基づいて、文書を予め決めておきます。その上で、開発系の文書に対して、検証業務を行う文書を定めておきます。
■苦い経験からの教訓
これらの文書一覧は、「標準的な文書一覧」として社内でリスト化・共有化しておくことで、誰でも同じ文書を作成出来るようになります。しかし実際のケースでは、供給者がGAMP・GMP・バリデーション・CSV…を理解していないケースがあります。その場合、予め供給者にどんな文書を作成して欲しいか?見積依頼時に説明しておくことで、供給者が作成・納品する文書に抜け漏れが無くなり、必要な文書作成工数や金額を見積もることが出来ます。
これまでの苦い経験として、供給者に文書作成とエビデンス取得、供給者内でのチェックの必要性を見積前、PJキックオフ時、各工程がスタートする前に何度も説明しないと、検証すべき文書が無かったり、不備が合ったりすることが多々ありました。結果として、その都度供給者に文書の作成/修正を依頼することになり、検証する側も供給者も想定以上の労力が発生してしまうため、個人的には重要な「儀式」だと考えています。
■文書を制する者はCSVを制する(?!)
以上、いかがでしたでしょうか?今回は「CSVの対象範囲はどこまで考えれば良いか?」という観点から、「CSV活動で必要となる文書」に着目して解説させて頂きました。CSV活動を含め、バリデーションにおいて「文書」という存在は、GxP対象業務及び使用される設備などの信頼性が保証されているか?確認・証明するために重要なものです。
どんな文書が必要なのか?を理解することは、どんなCSV活動があるのか?を理解することと同様です。是非、今回ご紹介した文書一覧例と解説を通じて、CSV活動上のお悩みが少しでも解消出来れば幸いです。
尚、上記のご質問やお悩みについて、2021年2月に「さらに理解が進む!応用できる!事例とQ&Aに基づくコンピュータ化システムバリデーション2021」と題したセミナーを開催しております。気になる方・会社様がおりましたら、情報機構様までお問い合わせ下さい。無料でご相談を承ります。
第4回「コンピュータ化システムバリデーションはどこまで実施すれば良いか?」(2021/9/10)
今回は、CSVのセミナーやアドバイスの中で一番多いご質問が多い「コンピュータ化システムバリデーション(CSV)はどこまで実施すれば良いか?」について、数回に分けて皆さんと一緒に学んで行きたいと思います。
このご質問は、特にCSVを実際に担当している方や、CSV対象PJが変更になると、必ず出てくる内容です。「どこまで実施すれば良いか?」という点は、CSV対象となるコンピュータ化システムや業務の特性に依存するため、唯一の答えはありません。しかし、1点だけ共通しているポイントがあります。それは「判断基準を何か?その理由は何か?」ということです。この判断基準はCSV担当者やシステムの特性によって変わるため、形式知化することを難しくしています。しかし、ポイントを押さえてしまえば、決して難しいものではありません。
早速「CSVをどこまで実施すれば良いか?」「その判断基準は何か?」という点について、皆様から頂くご質問やお悩みを3種類の定義で整理してみましょう(以下例です)。
①CSVの対象範囲
・新規導入/再構築/機能追加するコンピュータ化システムと関連するシステムの範囲は?
・CSVを実施するにあたり、ガイドライン/ERES/DIがどこまで関係するのか?
・DQ/IQ/OQ/PQ実施の対象範囲は?(特にExcelやAccess、クラウドサービス)
・CSVで必要となる文書として、何をどこまで作成すれば良いか?
・CSV担当者として、検証業務にどこまで関われば良いか?
② CSV実施の深さ/粒度
・サプライヤアセスメントやリスクアセスメントは、どこまで掘り下げて実施すべきか?
・CSVに関係するガイドライン/ERES/DIについて、どこまで深く理解しておくべきか?
・適格性評価にあたり、必要最低限必要なチェック項目(プロトコル)はどこまでか?
・作成するCSV関連文書の内容は、どんな粒度で記載すれば良いか?
・サプライヤが実施した作業に対し、どこまで検証すれば合格と評価出来るのか?
③CSV実施の判断基準
・コンピュータ化システムと関連するシステムの範囲をどのように決めれば良いか?
・カテゴリ分類を決める際の判断基準は?(特にスプレッドシートやPLC)
・CSV実施にあたりERES/DIがどこまで関わるか、どのように判断すれば良いか?
・DQ/IQ/OQ/PQ実施有無の明確な判断基準は何か?
・クラウドサービスを利用しても大丈夫かどうか?評価するための判断基準は?
これらは、CSV実施の現場で実際に見てきた内容やこれまでのご質問の一部ですが、必ずこれからの悩みや壁にぶつかります。そういった際に、過去PJの資料を漁ったり、CSVの基礎セミナーを受講していることと思います。対処例の1つとして、公開されている査察時の指摘事項とその理由を読むことも強くオススメします。特に、上記③判断基準を知る上でのポイントが詰まっているためです。
次回以降、「①CSVの対象範囲」「②CSV実施の深さ/粒度」「③CSV実施の判断基準」について、それぞれ事例を1つ取り上げて解説していきます。また、先行して、当方のブログにて9月中旬に「①CSVの対象範囲」のお悩みの1つをピックアップして、解説したいと思います。
尚、上記のご質問やお悩みについて、2021年2月に「さらに理解が進む!応用できる!事例とQ&Aに基づくコンピュータ化システムバリデーション2021」と題したセミナーを開催しております。気になる方・会社様がおりましたら、情報機構様までお問い合わせ下さい。無料でご相談を承ります。
☆★ 新規CSVのLMSを開講します!★☆
これからCSVを学ぶ方や、自社に足りない活動要素を補完したいとお考えの担当者様に、「ケーススタディで身に付けるCSV(コンピュータ化システムバリデーション)実践講座 (仮)」を開講する予定です。本講座の特長は以下3点です。
・新任のCSV担当者に最低限必要な基礎知識、最新の各種ガイドラインを「知る」
・改正GMP省令を踏まえたCSV活動の内容と流れが「分かる」
・ここまで学んだCSVの知識と活動内容を基に、ケーススタディを通じてCSV活動を「体験する(思考する)」
本講座はの目的は「基礎知識+実践」を組み合わせ、CSV活動の疑似体験頂くことで基礎知識と実務の流れを定着化することです。また、CSV活動における悩みやすいポイントに対して、補足資料や解説を加えながら実務に活かせる構成となっております。詳細に関しまして、後日改めて掲載させて頂きます。
第3回「コンピュータシステムとコンピュータ化システムの違いは何か?」(2021/8/4)
今回は、CSVの中でも誤解しやすいワードである「コンピュータシステム」と「コンピュータ化システム」の違いについて、皆さんと一緒に学んで行きたいと思います。
これは意外かもしれませんが、製薬/医薬品製造メーカーのCSV担当者も誤解していたり、目的と手段が反対になっていることが多いです(サプライヤーであるITベンダーは、さらに理解していません)。
このポイントを見誤ると「なぜこんなシステムを作ってしまったのか…」となり、GMP対象業務プロセスに対して、様々な問題やリスクを含んだまま業務を進めることになってしまいます。冒頭の2つの違いについて、自社に導入されているシステムや、導入検討中のシステムを想像しながら、考えてみましょう。
1.用語の定義
2つの用語の差である「化」という文字が入ることで、混乱するケースが多いようです。それぞれの言葉の定義とその差異を理解し、そこからCSVの本質を理解して行きましょう。以下に2つの用語の定義を示します。最大の違いは何か?なぜ違うのか?、3分程考えてみましょう。特に、CSV初心者の方は、CSVに精通している先輩社員が居れば、自分の考えを言葉で伝えて見て下さい。
① コ ン ピュ ー タ シ ス テ ム
特定の機能又は一連の機能を実行するために、設計し、組み立てられたハードウェア
及び関連するソフトウェアのグループのこと。
② コ ン ピュ ー タ 化 シ ス テ ム
コンピュータシステムで統合された工程又は作業、及びコンピュータシステムにより
実現される機能を利用する業務プロセスのこと。
さて、最大の違いは何でしょうか?一言で言うと「システム自体と、システム機能を利用した業務プロセス」の違いです。特に「業務プロセスまで検証し、文書化することがコンピュータ化システム(CSV)の重要な点」です(第2回のコラムを参照)。
2.望ましくないコンピュータ化システムの一例
それでは、この両者の違いを知らないまま、GMP対象業務にシステムシステムを導入するPJが立ち上がったと想定してみましょう。以下は、筆者が実際に実施/推進/アドバイスしてきた一例ですが、実際に現場で起きていることです。自社でも同じようなことが起きていないか?という目で見てみましょう。
<CSV担当者>
・ 「コンピュータシステムバリデーション」と捉えてしまうため、本来システム導入対象の業務と部門に対して、どのような文書を作成すれば良いが?/どのような内容を記載すれば良いか?提言出来ない(特に、URSに「要件」を書くことを業務部門に教えることが出来ない)。
・ 「コンピュータシステム」をバリデーションすることと誤解し、導入したシステムの検証だけに留まってしまう(ただのテストと変わりが無くなる)。
・ 本来のCSVであるコンピュータ化システムを通じて整備された業務プロセスから、正しい情報が得られるような検証方法/項目を検討することが出来ない。
<業務部門>
・ URSを作成するにあたり「要求仕様」ではなく「必要機能」を記載してしまい、業務全体として何を達成したいのか?不明確なURSを作成してしまう。
・ 上記URSを基にしてサプライヤー向けににRFPを発行する際、どんな機能が欲しいか?中心に提案を求めてしまう(システム導入が目的化してしまう)。
・ システム稼働後、本来の業務に必要な機能や不具合が見つかり、逸脱報告とシステム修正変更が相次ぐ(この頃には「なぜこんなシステムを作ってしまったのか…」となる)。
<サプライヤ>
・ 業務部門が必要とする機能の提案が中心となり、アプリケーションや機器類のプログラムのカスタマイズ/アドオンが多くなる。
・ 無駄な作り込みと複雑な仕様となるため、開発/保守コストが高くなる。
・ (システム管理業務を委託している場合)システム稼働後、システム変更に追われ、システム運用管理上本来必要な作業内容記録・設定書の変更・エビデンスの取得・変更管理の記録、文書管理が疎かになる。その上、自己点検が行われず、問題に気づかない。
3.本来のコンピュータ化システムと今求められる対応
ここまで「コンピュータシステム」と「コンピュータ化システム」の違いと、その違いから生まれる現実(望ましくない現状)を見てきました。たった一文字の違いの違いですが、求められるCSV活動は全く別物です。
第2回のコラムでも解説した通りですが、CSV担当者は自分の業務を通じて、信頼出来る製造業務プロセスを確立し、不良品が無い医薬品を人々に届け、「人々が健康である」状態にすることが真の目的です。その上で、今年8月から施行される改正GMP省令への対応、バリデーションへの影響、品質管理文化の醸成・品質保証部の設立・リスクベースアプローチなどへの対応が求められています。
加えて、海外のガイドラインや流れに合わせて、DIやデータ管理、外部のXaaSを利用する際のクオリフィケーション…その他、何からどのように手をつければ良いか?その計画立案と実装に苦労しているかと思います。もちろん、最初から全てを満足することは出来ません。あくまで、今後のブループリント(仮)を描きつつ、一つずつ現場の理解を得ながら進めていくことが正攻法です。
また、今回のコラムを補足する内容として、「2.望ましくないコンピュータ化システムの一例」を少しでも解消する方法を当方のブログで公開いたします。少し珍しい視点として「他業種」での取り組みから考察/共有したいと思います。
第2回「そもそも、なぜコンピュータ化システムバリデーション(CSV)を行うのか」(2021/7/5)
今回は「CSVの目的」「CSVの考え方」「真の目的」について、皆さんと一緒に学んで行きたいと思います。ポイントは「なぜCSVを行うのか?」「CSVの先に何があるのか?」…これらを押さえることで、CSV活動、人財育成、品質管理・保証の在り方、存在意義を会社全体で共通認識・継続的改善が出来るようになります。是非、社内の方と閲覧/議論してみて下さい。
1.なぜCSVを行うのか?
CSV活動の背景として、医薬品は生命に直接関係するため、価格に関係なく不良品ゼロが求められます。これはどの製薬・医薬品製造会社においても共通する点です。
しかし、実際には以下の背景から、製造会社は「医薬品製造に関わるコンピュータが目的通りに正常に動作すること」、「品質に適合する医薬品を恒常的に製造できることを実現すること」が命題となります。
● 医薬品産業におけるコンピュータの利用が近年ますます増加し、従来、人が行っていた多くの作業をコンピュータが行うようになった。
● 一方、コンピュータの欠陥による重大な事故も発生し、コンピュータシステムの信頼性と安全性が求められることになった。
● 上記の信頼性と安全性に対する最終責任は供給者(ITベンダー)ではなく、医薬品メーカーにある。そのため、コンピュータシステムの中身を理解する必要がある。
2.CSVの考え方とは?
前述の背景及び企業命題に対して、私達が取り組む活動がCSVです。CSVの考え方は、下図の通りとなります。

ここで最も重要な点は、CSVを通じて信頼性を確保するためには、「正しく運用するため、業務プロセスも含めて保証する」ことです。CSVは、コンピュータ化システムが期待通りに開発・設計・テストされ、それらを検証すること…だと思われがちですが、システムは製造工程上のツールであり、本来保証すべき範囲はGMP対象となる業務プロセスなのです。この業務プロセスに問題や不備があると、冒頭の「品質に適合する医薬品を恒常的に製造できる」とは言えないリスクを含むことなってしまいます。
3.真の目的は何か?
ここまでCSVの目的と考え方について解説してきました。さて、ここで読者の皆様に「質問」です。次の内容を1分程度、想像してみて下さい。又は、部署内で議論してみましょう。
「CSVを実施したら、どんな良いことがありますか?」
読者の皆さんは、どのようなことを考えたでしょうか?例えば、以下のようなことが思い付いた方が多いかもしれません(当方の推測です)。
● 製薬・医薬品製造において、不良品の発生率がゼロに近づく。
● どんな工場の要員でも、安心・安全な医薬品を製造出来るようになる。
● 査察官の抜き打ちチェックを受けても、信頼性出来る製造工程であると認められる。
上記内容は正しいですが、局所的な目的と言えます。それでは、もう少し掘り下げて考えるため、皆様の会社のホームページにある「企業理念」を一度見てみましょう。各社の企業理念に共通していそうなキーワードは「人々の健康で豊かな生活」ではないでしょうか?
つまり、CSV担当者は自分の業務を通じて、信頼出来る製造業務プロセスを確立し、不良品が無い医薬品を人々に届け、「人々が健康である」状態にすることが真の目的と言えるのではないでしょうか。各部署や経営陣は、この目標に向かって企業活動を行っているのです。
尚、今回のコラムを補足する内容を当方ブログで公開いたしますので、CSV活動に必要な考え方・姿勢・世界的な潮流について、一緒により深く学んでいきましょう。
北澤先生のブログはこちら(https://ameblo.jp/cocolo-smiling/)
第1回「自己紹介とCSV担当者の悩み」(2021/6/1)
■自己紹介
皆さん、はじめまして、CSVアドバイザーの北澤祐弥と申します。これまで、情報機構様を通じて「CoCoLoスマイリング」の北澤祐弥として、CSV関連セミナーを開催させて頂いておりました。
その上で、改正GMP省令や海外の潮流を踏まえたCSV活動の在り方・実践的な取り組み方を研究し、CSVが取り組みやすくなるサービスを提供する「C&Gファーマサポート」を2021年に3月に立ち上げさせて頂きました(設立の経緯はコチラをご覧下さい)。
今後、本コラム及びセミナーを通じて、皆さんと一緒にCSVに対する理解を深めていきたいと思いますので、どうぞ宜しくお願いいたします。
■CSV担当者の悩み
製薬・医薬品製造業界において「コンピュータ化システム」と「コンピュータシステム」の違いは、天と地の差があります。品質管理部や品質保証部などに配属・異動し、初めてCSVに取り組む方は、まずこの違いを理解するところからスタートします。
また、製薬・医薬品製造会社にシステム導入を行うITベンダー(サプライヤ)はCSV自体を理解していないことが多く、コンピュータ化システムとコンピュータシステムの違いを理解しないままに、システム導入を行おうとします。しかし、実際には当初考えていたシステム導入と異なる「検証業務」という活動の存在に悩みます。
そういったCSVに初めて関わる担当者は、「コンピュータ化システムバリデーション入門編」のようなセミナーの受講、及び書籍「コンピュータ化システム適正管理ガイドライン入門(株式会社じほう)」を通じて基本を学ぼうとしますが、いざ活動してみるとなかなか難しいのが現実です。実際に、現在CSV活動に従事している方のセミナー参加や、実務での悩みやご質問を頂くことが多くございます。
■本当に使えるCSV知識
本コラムは皆さんが一番理解したい内容・教えて欲しい内容から、CSV活動の基本と活動内容を丁寧に紐解く形で進めていきたいと思いますので、是非教えて欲しいことやお悩みなどありましたら、ご連絡頂ければ幸いです。
それでは、次回「そもそも、なぜコンピュータ化システムバリデーションを行うのか(仮)」をテーマにコラムをスタートいたしますので、気長にお付き合い下さい。
■おまけ 情報機構セミナー企画担当からひとこと
情報機構として、久しぶりの講師コラム&初の「CSV」関連コラムがスタートしました!
ご執筆を担当いただく北澤先生のCSVセミナーは、資料も大変分かりやすく、解説も質疑対応もとても丁寧、と定評をいただいております。企画担当としても自信をもってオススメ出来るセミナーです。
CSV活動について、日々困っている…何を参考にすれば良いのかわからない…そんな皆様へ、普段のご業務の合間にお気軽に読んで頂ける「CSV活動のあるある」「最近のトピック」を寄せ集めたコラムです。どうぞご期待ください!
|